Chapter 11. Heart
(Atlas of Ultrasound in Ob and Gyn)
1. Overview of Congenital Heart Disease
Congenital heart disease affects between 8 and 12 per 1,000 live births and 27 per 1,000 stillbirths. The risk of congenital heart disease increases if one parent has congenital heart disease, or if the couple has had a previous child with congenital heart disease. Risks are also increased with various genetic syndromes, both dominant and recessive, and with aneuploidy. Approximately 5% of infants born with cardiac anomalies have a positive family history of congenital heart disease, and approximately 12% have abnormal chromosomes.Prenatal diagnosis of congenital heart disease can improve outcome by leading to timely management and treatment after birth. Another avenue to improved outcome is the use of fetal interventions for some cardiac anomalies, where in utero balloon dilation procedures or stent placement can prevent worsening of the cardiac abnormality as pregnancy progresses (e.g., aortic stenosis dilation to prevent or minimize development of hypoplastic left heart).
2. Hypoplastic Left Heart Syndrome and Aortic Stenosis
DESCRIPTION AND CLINICAL FEATURES
Hypoplastic left heart syndrome is a group of cardiac malformations characterized by a small or absent left ventricle. Hypoplasia of the left ventricle occurs as a result of abnormalities that limit the flow of blood through the left side of the heart, including stenosis or atresia of the foramen ovale, mitral valve, or aortic valve. Aortic stenosis or atresia most commonly results from an abnormal aortic valve, but occasionally, the obstruction occurs in the subaortic region of the left ventricular outflow tract or beyond the valve. Not all cases of aortic stenosis or atresia lead to hypoplasia of the left ventricle. In particular, in some cases of aortic stenosis or atresia, a ventricular septal defect is present, allowing enough blood to flow through the left ventricle to prevent the development of a hypoplastic left ventricle. Even in cases of aortic stenosis or atresia without a ventricular septal defect, the left ventricle may not become hypoplastic initially but may instead dilate and develop endocardial fibroelastosis, becoming progressively less contractile. Eventually, however, the dilated, globular, poorly contractile left ventricle will shrink and become hypoplastic.
In general, the prognosis for a fetus with hypoplastic left ventricle is poor. Fetuses may develop hydrops in utero and may die before birth. For infants with hypoplastic left heart born alive, survival has improved in the last few decades as a result of the development of palliative surgical interventions and cardiac transplantation.
The prognosis for aortic stenosis is related to the degree of function of the left cardiac ventricle.
DESCRIPTION AND CLINICAL FEATURES
Hypoplastic left heart syndrome is a group of cardiac malformations characterized by a small or absent left ventricle. Hypoplasia of the left ventricle occurs as a result of abnormalities that limit the flow of blood through the left side of the heart, including stenosis or atresia of the foramen ovale, mitral valve, or aortic valve. Aortic stenosis or atresia most commonly results from an abnormal aortic valve, but occasionally, the obstruction occurs in the subaortic region of the left ventricular outflow tract or beyond the valve. Not all cases of aortic stenosis or atresia lead to hypoplasia of the left ventricle. In particular, in some cases of aortic stenosis or atresia, a ventricular septal defect is present, allowing enough blood to flow through the left ventricle to prevent the development of a hypoplastic left ventricle. Even in cases of aortic stenosis or atresia without a ventricular septal defect, the left ventricle may not become hypoplastic initially but may instead dilate and develop endocardial fibroelastosis, becoming progressively less contractile. Eventually, however, the dilated, globular, poorly contractile left ventricle will shrink and become hypoplastic.
In general, the prognosis for a fetus with hypoplastic left ventricle is poor. Fetuses may develop hydrops in utero and may die before birth. For infants with hypoplastic left heart born alive, survival has improved in the last few decades as a result of the development of palliative surgical interventions and cardiac transplantation.
The prognosis for aortic stenosis is related to the degree of function of the left cardiac ventricle.

 |
| FIGURE 11.2.2 Hypoplastic left ventricle with no left ventricular chamber. A: Transverse image of thorax demonstrating heart with large right ventricle (RV, arrow) and no appreciable left ventricular chamber (LV, arrow). The left atrium (LA, arrowhead) is also small compared to the right atrium (RA, arrowhead). B: Video clip of same heart beating showing no appreciable left ventricular chamber.
|
SONOGRAPHY
A hypoplastic left ventricle can be diagnosed on a four-chamber view of the heart by demonstrating a small left ventricle (Figure 11.2.1), often with poor contractility. The size of the left ventricular chamber is variable. In some cases, no left ventricle can be identified (Figure 11.2.2). In others, the left ventricle is only somewhat smaller than the right. With ventricular dilatation and endocardial fibroelastosis as a result of severe aortic stenosis, the left ventricle becomes enlarged and globular, with increased echogenicity along the inner wall and poor ventricular contractility (Figure 11.2.3). As pregnancy progresses, the dilated, globular left ventricle may become progressively smaller in utero until it becomes hypoplastic. The increased echogenicity of the ventricular wall will persist (Figure 11.2.4).


Aortic stenosis is characterized by narrowing of the aortic valve and decreased movement of the valve leaflets. The narrowed aortic valve can be visualized on a long-axis view of the left ventricle and left ventricular outflow tract (Figure 11.2.5). The width of the valve can be measured and compared with norms for gestational age. The stenotic valve is often brightly echogenic and visible throughout the cardiac cycle. The ascending aorta may be enlarged due to poststenotic dilation. Color Doppler will demonstrate a narrow, high-velocity jet of flow across the stenotic valve (Figure 11.2.6), instead of the normal broad area of flow.

 |
| FIGURE 11.2.6 Aortic stenosis and mitral regurgitation with color Doppler. A: Oblique color Doppler image showing left ventricular (LV) outflow tract with narrowing of the color jet (large arrow) and aliasing of the color signal, indicating high-velocity flow across the stenotic valve. The ascending aorta is dilated (arrowhead) due to poststenotic turbulence above the valve. Also seen is a large jet of retrograde flow across the mitral valve (small arrow), representing mitral regurgitation, which often accompanies critical aortic stenosis. B: Another fetus with similar findings of a narrowed aortic jet (arrowhead) across the stenotic valve and large jet of retrograde flow across the mitral valve (arrow). C and D: Video clips with color Doppler in fetuses (A) and (B), respectively showing the narrowed jet of flow across the aortic valve and the large jet of mitral regurgitation.
|
Mitral regurgitation commonly accompanies aortic stenosis when hypoplastic left heart is developing. With color Doppler imaging, the regurgitation appears as retrograde flow across the mitral valve during cardiac systole (Figure 11.2.6).
3. Hypoplastic Right Ventricle and Pulmonic Stenosis
DESCRIPTION AND CLINICAL FEATURES
A hypoplastic right ventricle, which is less common than hypoplastic left heart, is characterized by a small or absent right cardiac ventricle. This most often results from pulmonic stenosis or atresia with an intact ventricular septum but can also result from stenosis or atresia of the tricuspid valve. With any of these, there is obstruction of blood flow either into the right ventricle or out of the right ventricle, leading to shunting of blood across the foramen ovale to the left side of the heart. The left ventricle may be enlarged and hypertrophic. A hypoplastic right ventricle may cause fetal cardiac failure and hydrops.
Pulmonic stenosis is characterized by an abnormal pulmonic valve that obstructs the blood flow through the right ventricular outflow tract. The stenosis may be an isolated abnormality of the heart or a component of a more complex congenital cardiac malformation such as tetralogy of Fallot. With isolated pulmonic stenosis, the right ventricle may be small or large depending on the degree of shunting of blood across the foramen ovale and the extent of tricuspid regurgitation.
SONOGRAPHY
A hypoplastic right ventricle is best diagnosed on a four-chamber view of the heart when the right ventricle is smaller than the left ventricle (Figure 11.3.1). The small right ventricle often has thickened walls, sometimes markedly (Figure 11.3.2), and ventricular contractility is usually poor. In rare cases, no right ventricle can be found (Figure 11.3.3). In the latter case, the distinction between hypoplastic right ventricle and hypoplastic left ventricle may be difficult.
3. Hypoplastic Right Ventricle and Pulmonic Stenosis
DESCRIPTION AND CLINICAL FEATURES
A hypoplastic right ventricle, which is less common than hypoplastic left heart, is characterized by a small or absent right cardiac ventricle. This most often results from pulmonic stenosis or atresia with an intact ventricular septum but can also result from stenosis or atresia of the tricuspid valve. With any of these, there is obstruction of blood flow either into the right ventricle or out of the right ventricle, leading to shunting of blood across the foramen ovale to the left side of the heart. The left ventricle may be enlarged and hypertrophic. A hypoplastic right ventricle may cause fetal cardiac failure and hydrops.
Pulmonic stenosis is characterized by an abnormal pulmonic valve that obstructs the blood flow through the right ventricular outflow tract. The stenosis may be an isolated abnormality of the heart or a component of a more complex congenital cardiac malformation such as tetralogy of Fallot. With isolated pulmonic stenosis, the right ventricle may be small or large depending on the degree of shunting of blood across the foramen ovale and the extent of tricuspid regurgitation.
SONOGRAPHY
A hypoplastic right ventricle is best diagnosed on a four-chamber view of the heart when the right ventricle is smaller than the left ventricle (Figure 11.3.1). The small right ventricle often has thickened walls, sometimes markedly (Figure 11.3.2), and ventricular contractility is usually poor. In rare cases, no right ventricle can be found (Figure 11.3.3). In the latter case, the distinction between hypoplastic right ventricle and hypoplastic left ventricle may be difficult.


With pulmonic stenosis, there is narrowing at the level of the pulmonic valve (Figure 11.3.4). Measurement of the pulmonic valve can be compared with norms for gestational age to assess the degree of narrowing. Poststenotic dilation of the pulmonic artery may be seen in some cases of isolated pulmonic stenosis (Figure 11.3.5). With pulmonic atresia or critical pulmonic stenosis, retrograde flow may be seen in the ductus arteriosus, carrying blood from the aorta to the pulmonary arteries. The reversed flow in the ductus arteriosus is best seen on a transverse color Doppler image of both the ductal arch and the aortic arch and is diagnosed when flow in the ductal arch is in the opposite direction to flow in the aortic arch (Figure 11.3.6). Careful assessment for accompanying cardiac anomalies is warranted, especially looking for abnormalities of the tricuspid valve and ventricular septum.


 |
| FIGURE 11.3.5 Pulmonic stenosis with poststenotic dilatation of the pulmonary artery. Oblique image of right ventricular (RV, arrowhead) outflow tract demonstrating narrow pulmonic valve (calipers) with poststenotic dilatation of the main pulmonary artery (arrows) (Post, posterior).
|

Ebstein Anomaly
DESCRIPTION AND CLINICAL FEATURES
Ebstein anomaly is an anomaly that involves malformation and malposition of the tricuspid valve. The valve is displaced into the right ventricle and is dysplastic and incompetent, leading to tricuspid regurgitation and enlargement of the right atrium. During atrial systole, blood flows from the right atrium toward the apex of the right ventricle. During ventricular systole, the blood regurgitates from the portion of the right ventricle distal to the tricuspid valve, back across the dysplastic tricuspid valve into the right atrium. The right atrium can become markedly enlarged. Hydrops may develop in utero due to fetal cardiac failure. The prognosis for this cardiac anomaly, when diagnosed prenatally, is poor, with a mortality of 35% to 40%. Some fetuses die before birth and others die in the neonatal period. The prognosis is particularly poor if hydrops develops in utero or when there is pulmonary hypoplasia as a result of compression of the lungs by the enlarged heart. Long-term survivors of Ebstein anomaly often have persistent cardiac arrhythmias.
SONOGRAPHY
With Ebstein anomaly, the four-chamber view of the heart is abnormal. The heart is markedly enlarged, especially the right atrium, and the tricuspid valve is displaced toward the apex of the right ventricle (Figure 11.4.1). Tricuspid regurgitation can be demonstrated with color or spectral Doppler (Figure 11.4.2).


5. Ventricular Septal Defect
DESCRIPTION AND CLINICAL FEATURES
An opening in the muscular or membranous portion of the interventricular septum is called a ventricular septal defect. These defects may be small and clinically insignificant or quite large, causing significant shunting of blood across the defect. Some of the small defects close spontaneously after birth. Defects in the membranous portion of the septum are more common than those in the muscular septum and tend to be smaller. Prenatally, blood flow through the septal defect is typically from the right to the left ventricle. After birth, shunting across the defect changes to be from left to right due to changes in pressure in the cardiac ventricles. Ventricular septal defects may be isolated anomalies or part of a more complex cardiac malformation. Isolated defects have an excellent prognosis.
SONOGRAPHY
The diagnosis of a ventricular septal defect is made when a gap is seen in the septum between the right and left ventricles. Many ventricular septal defects can be diagnosed on a four-chamber view of the fetal heart (Figure 11.5.1). Some defects, such as some smaller defects in the membranous portion of the septum, will only be seen on a long-axis view of the left ventricle and left ventricular outflow tract (Figure 11.5.2). Other ventricular septal defects, especially very small membranous ones, may not be visible at all on prenatal ultrasound. Flow across ventricular septal defects in utero, typically from the right to the left ventricle, can be seen with color Doppler imaging (Figure 11.5.3). Because a ventricular septal defect may be a component of a complex cardiac anomaly, careful assessment of ventricular and atrial chamber sizes, atrioventricular valves, ventricular outflow tracts, and the atrial septum is warranted.



6. Atrioventricular Canal
DESCRIPTION AND CLINICAL FEATURES
Atrioventricular canal is a severe cardiac anomaly characterized by a large defect in the central portion of the heart. This part of the heart is sometimes referred to as the “endocardial cushion,” and hence atrioventricular canal is sometimes termed “endocardial cushion defect.” The defect involves both atrioventricular valves and both the atrial and ventricular septa. As a result, the atria and ventricles communicate across septal defects as well as between the atria and ventricles across the defects in the atrioventricular valves. Anomalous formation of the ventricular outflow tracts is common. Fetuses with this cardiac anomaly often have other structural anomalies or aneuploidy, particularly trisomy 21. The prognosis is usually poor.
SONOGRAPHY
On the four-chamber view of the heart, atrioventricular canal is evident as a large defect in the middle of the heart, with absence of portions of the atrial and ventricular septum and abnormal atrioventricular valves (Figure 11.6.1). The atrioventricular valves may appear as one large valve between the atria and ventricles with a straight or slightly convex shape toward the ventricles. The diagnosis of atrioventricular canal may be difficult to make when the valves are closed and much easier when the valves are open (Figure 11.6.2). Because of the association of atrioventricular canal with aneuploidy and other cardiac and noncardiac anomalies, a careful fetal anatomic survey should be performed to search for other anomalies.


DESCRIPTION AND CLINICAL FEATURES
Tetralogy of Fallot is a complex cardiac anomaly comprising four cardiac abnormalities in the neonate: pulmonic stenosis, ventricular septal defect, overriding aorta, and right ventricular hypertrophy. In the fetus with tetralogy of Fallot, only the first three components of this anomaly are present. In general, the prognosis is good for this cardiac malformation because corrective surgery can be performed after birth. Rarely will the fetus develop hydrops due to cardiac failure.
SONOGRAPHY
Fetuses with tetralogy of Fallot may have a normal four-chamber view of the heart, and the diagnosis may be missed if the ventricular outflow tracts are not carefully evaluated. Tetralogy of Fallot is best diagnosed on a long-axis view of the left ventricular outflow tract where the ventricular septal defect and overriding aorta are visible (Figure 11.7.1). “Overriding aorta” refers to widening of the aorta at the level of the aortic valve, such that the ascending aorta extends over the ventricular septal defect to overlie part of the right ventricle. Narrowing of the pulmonic valve can be demonstrated on either a transverse or longitudinal view of the right ventricular outflow tract (Figure 11.7.2). With tetralogy of Fallot, the pulmonary artery is typically smaller than the aorta, whereas the pulmonary artery is larger than the aorta in the normal heart. This altered relationship in size of the two great vessels is often well demonstrated on the three-vessel view (Figure 11.7.3).



8. Transposition of the Great Vessels
Transposition of the great vessels is characterized by reversed ventricular outflow tracts, with the pulmonary artery arising from the left ventricle and the aorta arising from the right ventricle. The anomaly is often accompanied by a ventricular septal defect. This cardiac defect has a fairly good prognosis, particularly when it is diagnosed prenatally because corrective surgery can be performed after birth.
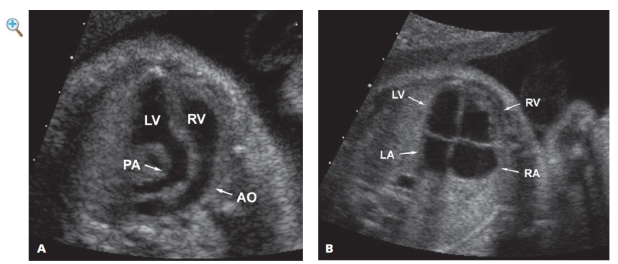 |
| FIGURE 11.8.1 Transposition of the great vessels. A: Image of outflow tracts demonstrating parallel configuration of the great vessels as they exit the heart, a configuration that indicates that the aorta (AO, small arrow) arises from the right ventricle (RV) and the pulmonary artery (PA, small arrow) arises from the left ventricle (LV). B: The four-chamber view of the heart is normal (RV small arrow, right ventricle; LV small arrow, left ventricle; RA small arrow, right atrium; LA small arrow, left atrium). C: Video clip through the beating heart showing normal-appearing four chambers and parallel configuration of the transposed great vessels. |
SONOGRAPHY
With transposition of the great vessels, the pulmonary artery and aorta arise from the base of the heart in parallel, as opposed to the crossed configuration of the normal outflow tracts. Because of the parallel configuration of the outflow tracts, the diagnosis is best made on a long-axis view of the left ventricle, which demonstrates both great vessels extending cephalad and parallel, the aorta anterior and to the right of the pulmonary artery (Figures 11.8.1 to 11.8.3). Identifying branching from the great vessel that arises from the left ventricle confirms that it is the pulmonary artery and not the aorta which has no branches at this level (Figures 11.8.2 and 11.8.3). Following the great vessel arising from the right ventricle will show that it forms the aortic arch with its branches and continues down the descending aorta (Figure 11.8.2). With transposition of the great vessels, the four-chamber view is usually normal unless either a ventricular septal defect is present or the transposition is a component of a more complex cardiac malformation, such as double-outlet right ventricle.



9. Truncus Arteriosus
DESCRIPTION AND CLINICAL FEATURES
With truncus arteriosus, a single large vessel arises from the base of the heart, receiving blood from both ventricles and branching to supply pulmonary, coronary, and systemic arteries. Truncus arteriosus is classified based on the type of branching of the truncus into the different arteries. This anomaly is typically a component of a complex congenital cardiac malformation, often involving hypoplasia of the left or right ventricle, a large ventricular septal defect, or an atrioventricular canal. The prognosis depends on the extent of the entire cardiac anomaly and, in general, is poor.
SONOGRAPHY
When truncus arteriosus is present, a single large vessel is seen arising from the base of the heart (Figure 11.9.1). Because of the association of truncus arteriosus with other anomalies of the heart, careful assessment of the cardiac ventricles and ventricular septum should be performed.

Double-Outlet Right Ventricle
DESCRIPTION AND CLINICAL FEATURES
Double-outlet right ventricle is a complex malformation of the heart in which both the aorta and the pulmonary artery arise from the right ventricle and no great vessel arises from the left ventricle. Often the aorta and pulmonary artery are transposed, with the aorta arising anterior and to the right of the pulmonary artery, aligned in parallel as they flow out of the right ventricle. Ventricular septal defects are commonly seen with this anomaly, so that blood entering the left ventricle is directed across the defect to the right ventricle before exiting the heart through the great vessels. Hypoplasia of the left ventricle is also common.
The prognosis with this anomaly, in general, is poor. Chromosomal abnormalities, especially trisomies 13 and 18 and DiGeorge syndrome (22q deletion), are common, affecting more than a third of cases. Noncardiac congenital anomalies are also frequently present. Overall, the prognosis depends on the associated anomalies in the heart, including the size of the ventricular septal defect and the sizes of the right and left ventricles, as well as the severity of associated noncardiac and chromosomal abnormalities.
SONOGRAPHY
The diagnosis of double-outlet right ventricle is made when both the pulmonary artery and the aorta arise from the right ventricle (Figure 11.10.1). Because the great vessels are typically transposed, the aorta and pulmonary artery will exit the heart in parallel, with the aorta anterior and to the right of the pulmonary artery. A ventricular septal defect is commonly present (Figure 11.10.1). Often the left ventricle is hypoplastic (Figure 11.10.2). Since this anomaly is commonly associated with other anomalies, careful sonographic assessment of fetal anatomy is warranted.


11. Myocardial Tumors
Tumors of the myocardium are most often rhabdomyomas, which, when multiple, are usually a manifestation of tuberous sclerosis. Rhabdomyomas are hamartomas of cardiac muscle that arise in the myocardium and tend to grow during gestation. Cardiac failure with hydrops in utero may result from either obstruction of blood flow by the rhabdomyoma or poor cardiac contractility due to replacement of normal myocardium by tumor. The prognosis for cardiac rhabdomyomas is related to the size and number of rhabdomyomas. If hydrops develops in utero, the prognosis is worse than in the absence of hydrops. After birth, cardiac rhabdomyomas occasionally regress.
SONOGRAPHY
Cardiac rhabdomyomas are visible as round or oval masses arising from the myocardium. They are typically more echogenic than normal cardiac muscle, and they may be single (Figure 11.11.1) or multiple (Figure 11.11.2). When hydrops is present, pericardial fluid may outline the myocardial tumors.


12. Arrhythmias
DESCRIPTION AND CLINICAL FEATURES
A variety of abnormal cardiac rhythms may be encountered in the fetus, including premature atrial contractions, bradycardia, atrioventricular heart block, and a number of tachyarrhythmias (e.g., supraventricular tachycardia, atrial flutter, and atrial fibrillation). In general, the prognosis is related to whether hydrops develops in utero. For some cardiac arrhythmias, antiarrhythmic drugs that cross the placenta can be given to the mother to correct the fetal arrhythmia. Premature atrial contractions occur commonly and are typically benign and self-limited. Fetuses with premature atrial contractions rarely develop hydrops or require medication in utero or after birth.

SONOGRAPHY
An abnormal cardiac rhythm can be seen with real-time sonography. M-mode sonography is used to document and characterize the arrhythmia because it can quantify atrial and ventricular rates separately. With bradycardia, the heart rate is abnormally slow, less than 110 beats per minute (bpm) in the second and third trimesters (Figure 11.12.1). With atrioventricular heart block, there is dissociation between the atrial beats and the ventricular beats, with the atria typically beating more rapidly than the ventricles (Figure 11.12.2). With supraventricular tachycardia, the heart rate is faster than normal, more than 180 bpm (Figure 11.12.3). If the tachycardia leads to hydrops, pericardial effusion, pleural effusions, ascites, and/or skin thickening will be present. With atrial flutter, the atrial rate is very fast, usually more than 300 bpm. The atrial rate is even faster (more than 400 bpm) with atrial fibrillation. With atrial flutter or fibrillation, the ventricular rate is usually slower than the atrial rate due to incomplete conduction (Figure 11.12.4).

Premature atrial contractions cause irregularity of the fetal heart rate characterized by an early contraction of the atria and ventricles followed by a pause before the normal rhythm resumes. Such premature atrial contractions may be infrequent or occasional or they may occur frequently. They can be observed in real time and documented by M-mode sonography (Figure 11.12.5).



13. Ectopia Cordis
DESCRIPTION AND CLINICAL FEATURES
Ectopia cordis is a rare anomaly in which the heart is located outside the fetal thorax, protruding through a defect in the anterior chest wall. In many cases, the heart is structurally abnormal in addition to being ectopically located. Although ectopia cordis may be an isolated anomaly, it is often a component of a syndrome such as pentalogy of Cantrell or amniotic bands. The prognosis is usually dismal.
SONOGRAPHY
When ectopia cordis is present, the heart is seen beating outside the chest of the fetus, extending from the thorax through a defect in the anterior thoracic wall (Figure 11.13.1). Pentalogy of Cantrell is diagnosed when there is ectopia cordis and an omphalocele (Figure 11.13.2). The other features of pentalogy of Cantrell include defects in the anterior diaphragm, the pericardium, and the inferior sternum, as well as cardiac anomalies.


14. Pericardial Effusion
Pericardial effusion is the abnormal accumulation of fluid between the visceral and parietal layers of the pericardium. The effusion may be isolated or a component of fetal hydrops. The presence of a trace amount of fluid carries little, if any, clinical significance, and small isolated effusions generally have an excellent prognosis. With a large effusion or one that is a component of hydrops, the prognosis is related to the underlying cause of the pericardial effusion or to the degree of hydrops. Large effusions can also be seen in fetuses with trisomy 21.

With ultrasound, a pericardial effusion is seen as a rim of fluid around the fetal heart (Figure 11.14.1). Care must be taken not to mistake the normal hypoechoic myocardium for pericardial fluid (Figure 11.14.2). The distinction can be made by noting that pericardial effusions change in shape with cardiac contractility and are completely anechoic. When the pericardial effusion is a component of hydrops, ascites, pleural effusions, and skin thickening may also be seen (Figure 11.14.3).


Pericardial Tumors
DESCRIPTION AND CLINICAL FEATURES
Tumors arising from the pericardium in utero are very rare. The most common is a pericardial teratoma, but pericardial hemangiomas and lipomas can also occur. Pericardial teratomas, like teratomas elsewhere, are benign germ cell tumors with histologic elements derived from the three germinal layers. Teratomas in the pericardium are almost always associated with a pericardial effusion. The prognosis for the fetus depends on the degree of cardiac tamponade by the tumor and/or the pericardial effusion. A poor prognostic sign is the development of hydrops in utero.
SONOGRAPHY
Pericardial teratomas appear sonographically as complex solid and cystic masses adjacent to the heart (Figure 11.15.1), often associated with pericardial effusion. The solid components may be very echogenic. If the tumor or pericardial effusion causes hydrops, ascites will be seen in the fetal abdomen (Figure 11.15.2).


Nhận xét
Đăng nhận xét