Chapter 15. Normal and Abnormal Fetal Anatomy
BS. Nguyễn Hồng Anh
Te ability to detect and characterize abnormalities beore birth is one o the marvels o modern obstetrics. Sonography can image the etus with remarkable precision. Components o the standard anatomic survey are listed in Table 15-1. Tese have been termed essential elements by the American College o Obstetricians and Gynecologists (2020) and minimal elements by the American Institute o Ultrasound in Medicine (2018). Ideally, residency training in Obstetrics and Gynecology includes education in acquisition and interpretation o these views. Resources provided by national societies assist with this endeavor (Abuhamad, 2018).
A detailed etal anatomic survey is a specialized examination that may include more than 60 components, each determined on a case-by-case basis. It is the primary method o evaluating at-risk pregnancies and characterizing etal abnormalities, thereby aiding consultation and delivery planning. Indications are shown in able 14-6 and discussed urther in Chapter 14 (p. 251). At Parkland hospital, the detailed survey includes, at minimum, an attempt to visualize all components required by the American Institute o Ultrasound in Medicine or normal detailed case submission (see able 15-1).
Tis chapter presents the standard and detailed anatomic surveys and some o the many etal abnormalities that may be detected when visualization is optimal. When imaging or measuring any etal structure, care should be taken to place the ocal zone at the appropriate level and to magniy the image appropriately (Abuhamad, 2018). Whenever a etal abnormality is identied, a detailed anatomic survey is recommended. With rare exception, amniocentesis with chromosomal microarray analysis also is oered. I a cardiac abnormality is identied, etal echocardiography is indicated. Additional indications or these specialized examinations and or etal magnetic resonance (MR) imaging are reviewed in Chapter 14.
BIOMETRY
In the rst trimester, crown-rump length (CRL) measurement
is used to establish or conrm gestational age (Appendix, p.
1234). Te earlier that sonography is perormed, the more
accurate this estimation. Gestational age assessment is reviewed
in Chapter 14 (p. 248). Te etus is imaged in the midsagittal
plane in a neutral, nonexed position so that its length can be
measured in a straight line (Fig. 15-1). Te average o three
measurements is used. First-trimester nuchal translucency measurement is reviewed in Chapter 14 (p. 249).
In the second and third trimesters, the biparietal diameter, head circumerence, abdominal circumerence, and emur length are measured to conrm gestational age, i not already established in the

TABLE 15-1. Components of Standard and Detailed Anatomic Surveys Standard Ultrasound Detailed Ultrasound, Additional Components
aIn addition to all standard anatomy components, these detailed ultrasound components are required by the American
Institute of Ultrasound in Medicine for normal cases submitted as part of the detailed ultrasound accreditation process. Modified from the American Institute of Ultrasound in Medicine, 2018, 2019, 2020a. rst trimester, and to estimate etal weight. Ultrasound equipment and report packages calculate these estimates using standardized nomograms (Hadlock, 1991). I an abnormality involving one o the parameters is suspected, consideration is given to excluding it rom the gestational age calculation. Fetal weight nomograms are available that do not include the head measurements or the emur measurement (Hadlock, 1984; Shepard, 1982).
Te biparietal diameter and head circumerence are measured in the transthalamic view. Tis is a transverse image that includes the midline alx cerebri, cavum septum pellucidum, thalami, and insula (Fig. 15-2A). Te cerebral hemispheres should appear symmetric, and the cerebellum should not be visible. Te biparietal diameter is measured perpendicular to the alx cerebri, rom the outer edge o the skull in the near eld to the inner edge o the skull in the ar eld. Te head circum- erence is measured by placing an ellipse around the outer edge o the skull. Te head circumerence may also be calculated by averaging the biparietal diameter and the occipito-rontal diameter and multiplying by π.
Te abdominal circumerence is measured in a transverse image that includes the stomach and the J-shaped conuence o the umbilical vein with the portal sinus. An ellipse is placed just outside the etal skin edge (Fig. 15-2B). Te image should appear as round as possible and ideally contain no more than 1 rib on either side. Te spine should be visible in cross-section at the 3 o’clock or 9 o’clock position, whereas the kidneys should not be visible, as they are lower in the abdomen. Abdominal circumerence is the biometric parameter most aected by etal growth. An abdominal circumerence below the 10th percentile may be used to diagnose etal-growth restriction (Chap. 47, p. 825).
Te emur length is measured with the ultrasound beam perpendicular to the long axis o the shat. Calipers are placed at each end o the calcied diaphysis (Fig. 15-2C). Troughout the second and third trimesters, the emur length to abdominal circumerence ratio is normally 20 to 24 percent. I this ratio is below 18 percent, a skeletal dysplasia should be considered, particularly i other long-bone measurements are lagging (p. 302). As discussed in Chapter 17 (p. 340), a mildly oreshortened emur length measurement is also a minor marker or
Down syndrome (Herrera, 2020b).
Various nomograms exist or other etal structures, including the transverse cerebellar diameter, ocular distances, nasal bone, ear length, jaw index, thoracic circumerence, and lengths o the liver, kidneys, long bones, and eet. Tey may be used to address specic questions regarding organ system abnormalities, congenital inection, or genetic syndromes (Appendix, pp. 1238–1241).

FIGURE 15-2 Normal biometry. A. Transthalamic view depicts measurement of the biparietal diameter (BPD) and head circumference (HC). Landmarks include the cavum septum pellucidum (CSP), thalami (T), and insula (I). B. Abdominal circumference view (AC) shows measurement and landmarks, which include the stomach (S) and the confluence of the umbilical vein (U) and the left portal vein. C. Femur length measurement (FL).

FIGURE 15-1 The crown-rump length measures 61 mm in this 12-week, 4-day fetus.
BRAIN AND SPINE
Standard sonographic evaluation o the etal brain includes three transverse (axial) views. As noted, the transthalamic view should contain the midline alx cerebri, cavum septum pellucidum (CSP), thalami, and insula (see Fig. 15-2A). Te CSP is the space between the two laminae that separate the rontal horns o the lateral ventricles. It should be visible between approximately 17 and 37 weeks’ gestation, but ater this, usion o the septi pellucidi may obliterate the cavum. Inability to visualize a normal CSP may indicate a midline brain abnormality (Fig. 15-3).
For example, the rontal horns are widely spaced apart in agenesis o the corpus callosum (ACC), whereas in cases o septo-optic dysplasia (de Morsier syndrome) and lobar holoprosencephaly, the rontal horns communicate. Discussed in Chapter 16 (p. 312), an abnormally wide CSP may also be ound with trisomy 18.
Te transventricular view lies superior to the transthalamic view and, as the name implies, includes the lateral ventricles. Te ventricles are measured at their atrium, which is the conuence o the temporal and occipital horns (Fig. 15-4).
Te measurement is normally 5 to 9 mm throughout the second and third trimesters. Cerebrospinal uid is produced within the ventricles by the choroid plexus. Choroid plexus cysts are present in 0.5 to 2 percent o uncomplicated pregnancies and approximately 30 to 50 percent o pregnancies with trisomy 18 (Fig. 16-5, p. 312) (Reddy, 2014).
A detailed ultrasound examination is generally oered when ound. In the absence o an associated ultrasound abnormality, or unless an aneuploidy screening test indicates increased risk or trisomy 18, a choroid plexus cyst is considered a normal variant. For the transcerebellar view, the transducer is angled back through the posterior ossa (Fig. 15-5). Structures visible in this image include the

FIGURE 15-3 Absence of the cavum septum pellucidum, with coronal (A) and transverse (B) images showing communication between the frontal horns (FH) of the lateral ventricles. This may be isolated but can occur in the setting of septo-optic dysplasia or lobar holoprosencephaly. C = choroid plexus; F = falx cerebri.
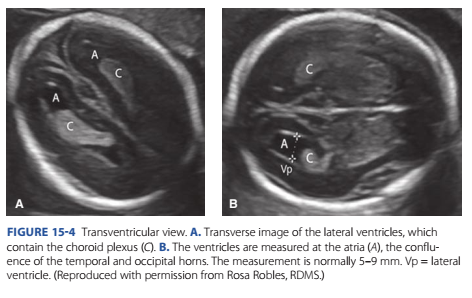
FIGURE 15-4 Transventricular view. A. Transverse image of the lateral ventricles, which contain the choroid plexus (C). B. The ventricles are measured at the atria (A), the confluence of the temporal and occipital horns. The measurement is normally 5–9 mm. Vp = lateral ventricle. (Reproduced with permission from Rosa Robles, RDMS.)
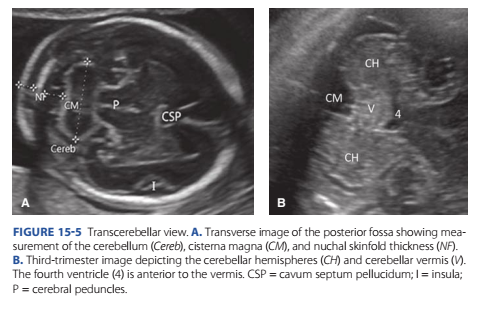
FIGURE 15-5 Transcerebellar view. A. Transverse image of the posterior fossa showing measurement of the cerebellum (Cereb), cisterna magna (CM), and nuchal skinfold thickness (NF). B. Third-trimester image depicting the cerebellar hemispheres (CH) and cerebellar vermis (V). The fourth ventricle (4) is anterior to the vermis. CSP = cavum septum pellucidum; I = insula; P = cerebral peduncles.
midline alx cerebri, cavum septum pellucidum, thalami, cerebellum, and cisterna magna. Te cerebellum and cisterna magna are measured, and between 15 and 20 weeks’ gestation, the nuchal skinold thickness also is measured. From 15 until 22 weeks, the cerebellar diameter in millimeters is roughly equivalent to the gestational age in weeks (Chavez, 2003). Cerebellar hypoplasia has been associated with various central nervous system (CNS) and non-CNS abnormalities (Howley, 2018). Te cisterna magna should measure between 2 and 10 mm throughout the second trimester and may reach 12 mm in the latter part o the third trimester. It becomes eaced when the Chiari II malormation is present (p. 277). I the cisterna magna is enlarged, the dierential diagnosis includes absence o all or part o the vermis (p. 279), a cyst such as an arachnoid cyst within the posterior ossa, or mega-cisterna magna, which is a diagnosis o exclusion and has an excellent prognosis. An increased nuchal skinold measurement is associated with increased risk or Down syndrome, other genetic syndromes, and structural abnormalities (Chap. 17, p. 341).
Imaging o the spine includes evaluation o the cervical, thoracic, lumbar, and sacral regions (Fig. 15-6). Representative images are oten obtained in the sagittal or coronal plane. However, imaging o each spinal segment in the transverse plane is more sensitive or anomaly detection. ransverse images demonstrate three ossication centers. Te anterior ossication center is the vertebral body, and the posterior paired ossication centers represent the junction o vertebral laminae and pedicles. Ossication o the spine proceeds in a cranial-caudal ashion. Te ossication o the upper sacrum (S1-S2) is not generally visible beore 16 weeks’ gestation, and ossication o the entire sacrum may not be visible until 21 weeks (De Biasio, 2003). Tus, detection o some spinal abnormalities can be challenging in the early second trimester.
■ Neuraltube Defects
Tese deects include anencephaly, myelomeningocele (spina bida), cephalocele, and rare spinal dysraphisms. Normally, the neural tube closes by the embryonic age o 26 to 28 days (Chap. 7, p. 124). Te birth prevalence o neural-tube deects approximates 0.9 in 1000 in the United States and most o Europe and 1.3 in 1000 in the United Kingdom (Cragan, 2009; Dolk, 2010). Many neural-tube deects can be prevented with olic acid supplementation. When isolated, neural-tube deect inheritance is multiactorial, and the recurrence risk without periconceptional olic acid supplementation is 3 to 5 percent (Chap. 16, p. 324).
Screening or neural-tube deects may be perormed with either ultrasound alone or ultrasound in addition to maternal serum alpha-etoprotein (MSAFP) level measurement (American College o Obstetricians and Gynecologists, 2019a). Between 15 and 20 weeks’ gestation, an upper MSAFP threshold o 2.5 multiples o the median (MoM) is anticipated to detect 95 percent with etal anencephaly and 80 percent with myelomeningocele. However, detection with standard sonography is at least comparable to that with MSAFP (Dashe, 2006; Norem 2005). A detailed ultrasound examination is the preerred diagnostic test and may identiy other abnormalities or conditions that also elevate MSAFP levels (able 17-5, p. 338).
Anencephaly is an absence o the cranium and telencephalon above the level o the skull base and orbits (Fig. 15-7). Acrania is absence o the cranium with protrusion o disorganized brain tissue, and herniation o the latter tissue is exencephaly. Importantly, brain tissue visible in the late rst trimester is oten not seen when ultrasound is perormed in the second or third trimester. Tus, anencephaly is the nal stage o exencephaly. It is oten diagnosed in the late rst trimester, and with adequate visualization, virtually all cases may be diagnosed in the second trimester.
Sonographically, the cranial contour may appear abnormal in the rst trimester and may resemble a “shower cap” in the late rst or early second trimester. Te ace oten appears triangular, and sagittal images demonstrate lack o an ossied cranium. Hydramnios rom impaired etal swallowing is common in the third trimester. Anencephaly is uniormly lethal. I the pregnancy is continued, perinatal palliative care consultation should be considered (American College o Obstetricians and Gynecologists, 2019b). Cephalocele is the herniation o meninges through a cranial deect, typically located in the midline occipital region (Fig. 15-8). When brain tissue herniates through the skull deect, the anomaly is termed an encephalocele. Herniation o the cerebellum and other posterior ossa structures constitutes a Chiari III malormation. Microcephaly is common by the third trimester. Associated intracranial abnormalities are requently visible, and survivors have a high incidence o neurological decits and intellectual disability. Cephalocele is an important eature o the autosomal recessive Meckel-Gruber syndrome, which includes cystic renal dysplasia and polydactyly. A cephalocele not located in the occipital midline raises suspicion or amnionic-band sequence (Chap. 6, p. 113).

FIGURE 15-6 Normal fetal spine. This sagittal image depicts the cervical (C), thoracic (T), lumbar (L), and sacral spine (S). Arrows denote the parallel rows of paired posterior ossification centers, which represent the junction of vertebral lamina and pedicles.
Spina bida is characterized by deects in the vertebrae, typically the dorsal arches, and subsequent exposure o the meninges and nerve roots. Te prevalence approximates 1 in 2000 births (Cragan, 2009; Dolk, 2010). Herniation o a meningeal sac and neural elements is a myelomeningocele (Fig. 15-9A). Less commonly, only an empty meningeal sac herniates, which is a meningocele. ransverse images are helpul to demonstrate separation or splaying o the lateral processes and characterize the level o the deect. Most cases are open spina bida, which means that the deect includes skin and sot tissues. Closed deects are skin-covered and more challenging to detect prenatally.
Detection o spina bida is aided by two characteristic cranial ndings (Nicolaides, 1986). Flatting or scalloping o the rontal bones is termed the “lemon sign,” and anterior curvature o the cerebellum with eacement o the cisterna magna is the “banana sign” (Fig. 15-9B,C). Tese ndings are maniestations o the Arnold-Chiari or Chiari II malormation. Tis develops when downward displacement o the spinal cord pulls a portion o the cerebellum through the oramen magnum and into the upper cervical canal. Te biparietal diameter measurement oten lags behind the other biometric parameters. Ventriculomegaly is common ater mid-gestation, and 80 to 90 percent o inants with myelomeningocele require ventriculoperitoneal

FIGURE 15-8 Encephalocele. This transverse image depicts a large defect in the occipital region of the cranium (arrows) through which meninges and brain tissue have herniated.

FIGURE 15-9 Myelomeningocele. A. Sagittal image of a lumbosacral myelomeningocele. Arrowheads indicate nerve roots within the anechoic herniated sac. The overlying skin abruptly stops at the defect (arrow). B. Transthalamic image demonstrating flattening of the frontal bones (arrows)—the lemon sign. C. Transcerebellar image depicting the banana sign, an anterior curvature of the cerebellum (arrows) and effacement of the cisterna magna.

FIGURE 15-7 Anencephaly/acrania. A. This transabdominal image at 11 weeks’ gestation depicts relatively subtle absence of the cranium. B. A transvaginal image at 11 weeks demonstrates more clearly the protrusion of a disorganized mass of brain tissue. C. By 14 weeks, this tissue resembles a “shower cap.” CRL = crown–rump length.
shunt placement (Adzick 2011, Chao, 2010). Aected children require multidisciplinary care to address problems related to the deect such as decits in swallowing, bladder and bowel unction, and ambulation. Fetal myelomeningocele surgery is discussed in Chapter 19 (p. 372).
■ Ventriculomegaly
Distention o the lateral ventricles is considered a nonspecic marker o abnormal brain development (Pilu, 2018). Mild ventriculomegaly is diagnosed when the atrial width measures 10 to 12 mm; moderate ventriculomegaly, when the measurement is 13 to 15 mm; and severe ventriculomegaly when >15 mm (Society or Maternal-Fetal Medicine, 2018). Representative images are depicted in Figure 15-10. Te choroid plexus may appear dangling in severe cases. Ventriculomegaly may occur secondary to a variety o central nervous system abnormalities and is associated with numerous genetic and inectious etiologies. Initial evaluation includes a detailed examination o etal anatomy, amniocentesis or chromosomal microarray analysis, and testing or congenital inections such as cytomegalovirus and toxoplasmosis (Chap. 16, p. 326). Genetic syndromes resulting in ventriculomegaly include L1 X-linked aqueductal stenosis, Joubert syndrome, Walker Warburg syndrome, hydrolethalus syndrome, and lissencephaly syndromes (Kousi, 2016). Ventriculomegaly does not typically result in signicant skull enlargement. wo exceptions—when macrocrania oten occurs—are aqueductal stenosis and the asymmetric ventriculomegaly interhemispheric cyst callosal dysgenesis (AVID) syndrome (Oh, 2019). Fetal MR imaging should be considered to assess or associated abnormalities that may not be detectable sonographically (Herrera, 2020a; Katz, 2018) (Chap. 14, p. 263).
Prognosis is determined by etiology, severity, and progression. In a systematic review o nearly 1500 mild to moderate cases, 1 to 2 percent were associated with congenital inection; 5 percent, with aneuploidy; and 12 percent, with neurological abnormality (Devaseelan, 2010). With chromosomal microarray analysis, genetic abnormalities may be identied in 10 to 15 percent (Society or Maternal-Fetal Medicine, 2018). Te larger the atria, the greater the likelihood o an underlying CNS abnormality and subsequent abnormal outcome (Gaglioti, 2009; Joó, 2008). I ventriculomegaly is isolated and remains mild, development is normal in at least 90 percent, and i moderate, development is normal in at least 75 percent (Society or Maternal-Fetal Medicine, 2018). Progression signicantly raises the likelihood o abnormal neurological development.
■ Agenesis of the Corpus Callosum
Te corpus callosum is a major ber bundle that connects reciprocal regions o the cerebral hemispheres. It is best

FIGURE 15-10 Ventriculomegaly. A. Mild ventriculomegaly. The atria measured 11 mm. No associated abnormality or underlying etiology was identified. B. Severe ventriculomegaly. In this fetus with aqueductal stenosis, the atria measured 45 mm. Arrow denotes the dangling choroid plexus.

FIGURE 15-11 Normal corpus callosum. A. Arrows point to the corpus callosum in this midsagittal image. B. Power Doppler image of the pericallosal artery (arrowheads).
l,viewed in the midsagittal plane, and color Doppler may demonstrate the pericallosal artery (Fig. 15-11). Agenesis o the corpus callosum has characteristic sonographic ndings. Te rontal horns are widely separated, and the occipital horns are rounded—which is called colpocephaly. ogether these ndings give the lateral ventricles a teardrop shape (Fig. 15-12). A normal cavum septum pellucidum is not visible because o rontal horn displacement. In the midline, bundles o Probst represent ber tracts that no longer cross in the midline. Ventriculomegaly is not uncommon, and without the corpus callosum bordering the third ventricle superiorly, the third ventricle may be elevated and mildly enlarged.
In population-based series, agenesis o the corpus callosum occurs in 1 in 4000 to 5000 pregnancies (Ballardini, 2018; Stoll, 2019; Szabo, 2011). It is associated with other anomalies, aneuploidy, and more than 200 genetic syndromes. Tus, chromosomal microarray analysis should be oered, and genetic counseling can be challenging. In a review o apparently isolated cases, etal MR imaging identied additional brain abnormalities in more than 20 percent o cases (Sotiriadis, 2012). I MR imaging does not identiy associated abnormalities, normal developmental outcome in 67 to 75 percent o cases and severe disability in about 10 percent has been reported (des Portes, 2018; Sotiriadis, 2012). other. Dierentiation into two cerebral hemispheres is induced by prechordal mesenchyme, which is also responsible or dierentiation o the midline ace. Cranioacial anomalies associated with holoprosencephaly are reviewed later (p. 283).
Te birth prevalence o holoprosencephaly is only 1 in 10,000. However, the abnormality has been identied in nearly 1 in 250 early abortuses, which attests to extremely high in-utero lethality (Orioli, 2010; Yamada, 2004). Te alobar orm accounts or 40 to 75 percent o cases, and 30 to 40 percent have a numerical chromosomal abnormality, particularly trisomy 13 (Orioli, 2010; Solomon, 2010). Conversely, o trisomy 13 cases, two thirds are ound to have holoprosencephaly.
■ DandyWalker Malformation
Tis posterior ossa abnormality is characterized by agenesis o the cerebellar vermis, posterior ossa enlargement, and elevation o the tentorium cerebelli. Te cerebellar hemispheres are visibly separated, and uid in the cisterna magna communicates with the ourth ventricle through the cerebellar vermis deect (Fig. 15-14). Te birth prevalence approximates 1 in 12,000 (Long, 2006). Associated anomalies and aneuploidy are common. Tese include ventriculomegaly in 30 to 40 percent, other anomalies in approximately 50 percent, and aneuploidy in 40 percent (Ecker, 2000; Long, 2006). Dandy-Walker malormation is also associated with numerous genetic and sporadic syndromes, congenital viral inections, and teratogen exposure, all o which greatly aect the prognosis. Tus, the initial evaluation mirrors that or ventriculomegaly (p. 278).
Inerior vermian agenesis is a term used when only the inerior portion o the vermis is absent. Even with partial and relatively subtle vermian agenesis, the prevalence o associated anomalies and aneuploidy is still high, and the prognosis is oten poor (Ecker, 2000; Long, 2006).

FIGURE 15-12 Agenesis of the corpus callosum. A. This transverse image demonstrates a teardrop-shaped ventricle. The frontal horns (F) are widely separated, no cavum septum pellucidum is visible, and bundles of Probst (P) line the midline. B. There is mild ventriculomegaly, no cavum septum pellucidum is visible (arrow), and the third ventricle (3V) is elevated and enlarged. A = atria.

FIGURE 15-13 Alobar holoprosencephaly. The thalami (Th) are fused and encircled by a monoventricle (V) with a covering mantle (M) of cortex. The midline falx is absent. (Reproduced with permission from Rafael Levy, RDMS.)

FIGURE 15-14 Dandy-Walker malformation. A. The cerebellar hemispheres (CH) are widely separated by a fluid collection that connects the 4th ventricle (4V) to the enlarged cisterna magna (CM). B. Sagittal image depicts elevation of the tentorium (arrows).
■ Holoprosencephaly
During early normal brain development, the prosencephalon or orebrain divides as it becomes the telencephalon and diencephalon. With holoprosencephaly, the prosencephalon ails to divide completely into two separate cerebral hemispheres and underlying paired diencephalic structures. Te most severe orm, alobar holoprosencephaly, is characterized by a single monoventricle that surrounds used thalami (Fig. 15-13). In semilobar holoprosencephaly, hemispheres partially separate. Lobar holoprosencephaly reers to a variable degree o usion o rontal structures and is more challenging to detect with prenatal ultrasound. Te lobar orm is among possible diagnoses when a normal cavum septum pellucidum cannot be visualized. Last, the middle interhemispheric variant o holoprosencephaly is characterized by communication between the midportion o the bodies o the lateral ventricles with separation o the rontal horns, such that the choroid plexus may prolapse rom one lateral ventricle into the
■ Schizencephaly and Porencephaly
Schizencephaly is a rare brain abnormality characterized by clets in one or both cerebral hemispheres, typically involving the perisylvian ssure. Te clet is lined by heterotopic gray matter and communicates with the ventricle, extending through the cortex to the pial surace (Fig. 15-15). Schizencephaly is believed to be an abnormality o neuronal migration, which explains its typically delayed recognition until ater midpregnancy (Howe, 2012). It is associated with absence o the cavum septum pellucidum, resulting in rontal horn communication. Ventriculomegaly is a common nding.
In contrast, porencephaly is a cystic space within the brain that is lined by white matter and may or may not communicate with the ventricular system. It is generally considered to be a destructive lesion. Porencephaly may develop ollowing intracranial hemorrhage in the setting o neonatal alloimmune thrombocytopenia or in an individual with a COL4A-1 mutation—a genetic condition which causes amilial porencephaly. In a monochorionic twin gestation, acute hypotension ollowing death o a co-twin also may create porencephaly. Fetal MR imaging should be considered when either o these CNS anomalies is identified.
■ Microcephaly
Tis condition indicates that the size o the head is prooundly smaller than expected. Te Society or Maternal-Fetal Medicine (2016) recommends that etal microcephaly be dened as a head circumerence at least 3 standard deviations (SD) below the mean or gestational age (Appendix, p. 1237). However, many etuses with measurements in this range have normal head size at birth, and thereore the diagnosis o pathologic microcephaly is not considered certain until the head circumerence reaches 5 SD below the mean (Society or Maternal-Fetal Medicine, 2016).
Te orehead is oten upsloping. Microcephaly is associated with a wide range o underlying abnormalities, genetic syndromes, and congenital inections such as toxoplasmosis, rubella, and cytomegalovirus, herpes, or Zika inection (Chap. 67, p. 1183). Findings that suggest inection include parenchymal and periventricular echogenic oci, ventriculomegaly, and cerebellar hypoplasia. Amniocentesis should be oered, and etal MR imaging should be considered. Te Society or Maternal-Fetal Medicine (2016) recommends perorming a detailed ultrasound examination o the etal brain i the head circumerence measures more than 2 SD below the mean. Another ultrasound evaluation ollows in 3 or 4 weeks, with the understanding that it may be difcult to distinguish constitutionally small head size rom pathologic ndings.
■ Sacrococcygeal Teratoma
Tis germ cell tumor is one o the most common tumors in neonates, with a birth prevalence o approximately 1 in 28,000 (Derikx, 2006; Swamy, 2008). It is thought to arise rom the totipotent cells along the Hensen node, anterior to the coccyx. Sacrococcygeal teratoma (SC) classication includes our types (Altman, 1974). ype 1 is predominantly external with a minimal presacral component; type 2 is predominantly external but with a signicant intrapelvic component; type 3 is predominantly internal and has abdominal extension; and type 4 is entirely internal with no external component. Te tumor histological type may be mature, immature, or malignant.
Sonographically, SC is a solid and/or cystic mass that arises rom the anterior sacrum and usually extends ineriorly and externally as it grows (Fig. 15-16). Solid components oten vary in echogenicity, appear disorganized, and may enlarge rapidly with advancing gestation. Fetal MR imaging should be considered because internal pelvic components may be challenging to visualize. Large, solid tumors requently result in hydrops due to high-output cardiac ailure, either as a consequence o tumor vascularity or secondary to bleeding within the tumor and resultant anemia. Hydramnios is common. Fetuses with tumors >5 cm oten require cesarean delivery, and classical hysterotomy may be needed (Gucciardo, 2011). Fetal therapy or SC is discussed in Chapter 19 (p. 373).
■ Caudal Regression Sequence
Tis rare anomaly is characterized by absence o the sacral spine and oten portions o the lumbar spine. It is approximately 25 times more prevalent in pregnancies complicated by diabetes (Garne, 2012). Caudal regression is associated with genitourinary malormations and syndromes such as the VACERL association (vertebral deects, anal atresia, cardiac deects, tracheo-esophageal stula, renal anomalies, and limb abnormalities) (Vilanova-Sanchez, 2018). Sonographic ndings include a spine that appears abnormally short, lacks normal lumbosacral curvature, and terminates abruptly above the level o the iliac wings (Fig. 15-17). Because the sacrum does not lie between the iliac wings, they are abnormally close together and may appear shield-like. Te lower extremities lack normal sot tissue development and may be abnormally positioned.
■ Sirenomelia
Tis rare anomaly may be conused with caudal regression sequence but is quite dierent. Formerly termed mermaid

FIGURE 15-15 Schizencephaly. This transverse image shows a large cleft extending from the right lateral ventricle through the cortex. Because the borders of the cleft are separate, the defect is termed open-lipped. (Reproduced with permission from Michael Davidson, RDMS.)
syndrome, it is characterized by a single lower extremity in the midline and bilateral renal agenesis. Te extremity may contain one or two sets o bones and eet (Fig. 15-18). Te bladder, anus, and genitalia are absent. What may appear to be a single umbilical artery in these cases is instead a remnant o the vitelline artery. Ater 18 weeks’ gestation, when the kidneys become the primary source o amnionic uid production, resultant anhydramnios may complicate the diagnosis. Cases o surviving inants have a variant o sirenomelia in which there is some unctioning renal tissue and urinary output (Pinette, 2005).
■ Hemivertebrae
Tese spinal segmentation-usion deects are characterized by abnormal development o hal o the vertebral
FIGURE 15-17 Caudal regression sequence. A. The spine is markedly foreshortened. The arrow shows where it terminates. B. The spine ends abruptly above the level of the iliac wings (I). C. Without a vertebral body between the iliac wings (I), they assume a shield shape.
FIGURE 15-18 Sirenomelia. A. This single lower extremity contained two femur bones (F), two lower leg bones (LL), and a fused foot with toes pointing outward (*). B. Arrows show the soft tissue outline of the lower extremity. Amnionic fluid is visible only because the gestational age is 17 weeks’ gestation. By 18 weeks, absence of kidneys and bladder resulted in anhydramnios. (Reproduced with permission from Melissa Salvie, RDMS.)

FIGURE 15-16 Sacrococcygeal teratoma. This tumor enlarged from 3 cm in diameter at 19 weeks’ gestation (A) to 9 cm in diameter during a 5-week period (B). Arrows depict the external borders of the mass.
body. Aected vertebrae are triangular and may be separate or used to adjacent vertebral bodies. Tis leads to abnormal curvature o the spine such as scoliosis (Fig. 15-19). Prenatally diagnosed cases typically involve the thoraco–lumbar spine. Associated abnormalities are common, particularly skeletal, renal, and cardiac deects (Basude, 2015; Yulia, 2020). Hemivertebrae are also a component o several syndromes, including the VACERL association.
HEAD, FACE, AND NECK
Cranioacial anatomy on the detailed anatomic survey may include cranial integrity and shape o the skull (Fig. 15-2A); images o the orbits, nose, and lips; views o the maxilla, mandible, hard palate, and tongue; images o the ears; and evaluation o the neck (Fig. 15-20). O these structures, only the upper lip is a component o the standard examination. At Parkland Hospital, we include an image o the sagittal prole as part o our standard examination to help detect micrognathia prenatally. When imaging the neck, particularly in the third trimester, identiying a nuchal cord is not uncommon (Fig. 15-21). Tis nding is not associated with adverse outcome, and we do not alter etal surveillance i a nuchal cord is detected.
A B
FIGURE 15-19 Hemivertebrae result in abnormal spinal curvature in these coronal images. (Reproduced with permission from Rose Muli, RDMS.)
A B C
D E F
FIGURE 15-20 Normal craniofacial and neck anatomy. A. Transverse images of orbits (O) and nose (N). The small circle within each orbit is the lens. The distance between the orbits roughly approximates the width of each orbit. B. Sagittal view of the face, depicting the nasal bone (NB), lips (L), maxilla (Max), and mandible (Man). C. Transverse image of the alveolar ridge. D. Coronal view of the nose, upper lip, and lower lip. E. Sagittal image of the neck. F. Image of the ear. (Reproduced with permission from Devi Nanandhan, RDMS.)
Hypertelorism (Fig. 15-23) is a common nding in trisomy 18. Hypotelorism, with or without microphthalmia, may be ound in the setting o holoprosencephaly. In its extreme orm only a single orbit is visible—cyclopia. Other comorbid anomalies with holoprosencephaly include a single nostril—cebocephaly, or absence o the entire nose (arhinia) with proboscis (Fig. 15-24).
Nasal bone hypoplasia or sonographic absence is an aneuploidy marker that coners increased risk or etal Down syndrome (Fig. 15-25). It is not a structural abnormality. Nasal bone measurement is part o the detailed ultrasound examination and is measured only between 15 and 22 weeks’ gestation. Te measurement is considered oreshortened i less than 2.5 mm or i more than 2 SD below the mean or gestational age (Chap. 17, p. 341). Hypoplasia o the nose is a dierent condition that can occur ollowing wararin exposure (Fig. 8-4, p. 156).
■ Facial Clefts
Tese are grouped into three main types. Te rst type, clet lip and palate, always involves the lip, may also involve the hard palate, can be unilateral or bilateral, and has a birth prevalence that approximates 1 in 1000 (Cragan, 2009; Dolk, 2010). I isolated, the inheritance is multiactorial—with a recurrence risk o 3 to 5 percent or one prior aected child. I a clet is visible in the upper lip, a transverse image at the level o the alveolar ridge may demonstrate that the deect also involves the primary palate (Fig. 15-26).
In one review o low-risk pregnancies, clet lip was identi- ed sonographically in only about hal o cases (Maarse, 2010). Approximately 40 percent o those detected prenatally are associated with other anomalies or syndromes, and aneuploidy is common (Maarse, 2011; Oerdal, 2008). Te rate o associated anomalies is highest or bilateral deects that involve the palate.
In the Utah Birth Deect Network, aneuploidy was identied in 1 percent with clet lip alone, 5 percent with unilateral clet lip and palate, and 13 percent with bilateral clet lip and palate (Walker, 2001).
Te second type o clet is isolated clet palate. It begins at the uvula, may involve the sot palate, and occasionally involves the hard palate—but does not involve the lip. Te birth prevalence approximates 1 in 2000 (Dolk, 2010). Identi- cation o an isolated clet palate has been described using detailed sonography, and particularly with threedimensional imaging, but detection is not easible in all cases (Ramos, 2010; Wilhelm, 2010). Iso lated clet palate is not expected to be visualized during a standard ultrasound examination (Maarse, 2011; Oerdal, 2008).
A third type o clet is median clet lip, which may
A B
FIGURE 15-22 Transthalamic images demonstrating dolichocephaly (A) and brachycephaly (B). The biparietal diameter (BPD) and head circumference (HC) are measured in each image.
FIGURE 15-21 Nuchal cord incidentally noted with color Doppler in a transverse image of the fetal neck at 34 weeks’ gestation.
■ Dolichocephaly and Brachycephaly
Te cephalic index reects the skull shape. It is measured by dividing the biparietal diameter by the occipitorontal diameter. Te cephalic index is normally 70 to 86 percent. It is smaller i the head shape is attened—dolichocephaly, and larger i the shape is rounded—brachycephaly. In such cases, the head circumerence measurement more reliably estimates gestational age than does the biparietal diameter (Fig. 15-22). Tese head shape variants may be normal or can be secondary to etal position or oligohydramnios. Dolichocephaly can occur with neural-tube deects, and brachycephaly may be seen in etuses with Down syndrome. A strawberry-shaped skull describes a pattern o angulation typical o trisomy 18 (Fig. 16-5, p. 312). With any abnormal skull shape, craniosynostosis is a consideration.
■ Abnormalities of Orbits and Nose
Subjectively, the distance in between the orbits is similar to the diameter o each orbit (see Fig. 15-20). Te lens is oten visible. Nomograms are available or the ocular diameter and or the interorbital and binocular distances (Appendix, p. 1240).
FIGURE 15-23 Abnormalities of the orbits. A. Hypertelorism in a fetus with trisomy 18. B. Hypotelorism in a fetus with trisomy 13 and alobar holoprosencephaly. C. Microphthalmia. This fetus also had trisomy 13. Arrows point to the eyes.
FIGURE 15-24 Nasal abnormalities associated with holoprosencephaly. A. Sagittal profile depicting a proboscis (arrow) protruding from
the forehead. B. Coronal image demonstrating the proboscis along with hypotelorism and absence of the nose. C. Coronal image demonstrating a single nostril (cebocephaly). D. Photograph of a newborn with cebocephaly.
FIGURE 15-25 Nasal bone (and its absence). A. Sagittal image of the profile showing measurement of a normal nasal bone at 19 weeks.
B. Fetus with trisomy 21, also at 19 weeks, with no visible nasal bone. (Reproduced with permission from Jason McWhirt, RDMS.)
be ound in etuses with holoprosencephaly, typically when hypotelorism is also present. Additionally, a median clet may be associated with hypertelorism and rontonasal dysplasia, ormerly called the median clet ace syndrome.
■ Micrognathia
Te etal prole can help identiy cases o micrognathia—hypoplasia o the mandible, and retrognathia, which is recession o the mandible in relation to the maxilla (Fig. 15-27). o quantiy micrognathia risk, a transverse image o the mandible can be used to calculate the jaw index, which is the anterior-posterior diameter o the mandible expressed as a percentage o the biparietal diameter (Paladini, 1999). Fetuses with micrognathia and retrognathia requently have a posterior clet palate and glossoptosis (recessed tongue)—a constellation o ndings known as the Pierre Robin sequence. Micrognathia is also a eature o reacher Collins syndrome, oral-acial-digital syndromes, trisomies 18 and 13, triploidy, and the 22 q11.2 deletion. Micrognathia may result in hydramnios and can cause airway obstruction at birth.
Use o the ex-utero intrapartum treatment (EXIT) procedure or severe micrognathia is discussed in Chapter 19 (p. 379). Micrognathia should not be conused with agnathia– otocephaly, a rare anomaly in which no mandible develops and the ears may use in the midline (Fig. 15-28). Te latter coners an extremely poor prognosis and has been diagnosed as early as the late rst trimester (Rodriguez, 2019).
■ Epignathus
Tis rare teratoma arises rom the oral cavity or pharynx. It may grow outward or both outward and into the brain, the latter conerring an extremely poor prognosis (Fig. 15-29). I brain involvement is absent, an EXI procedure, reviewed in Chapter 19 (p. 379), can help secure the airway at delivery (Chung, 2012).
■ Cystic Hygroma
Tis venolymphatic malormation is characterized by uid- lled sacs that extend rom the posterior neck (Fig. 15-30). Cystic hygromas may be diagnosed in the rst trimester and vary widely in size. Impaired lymphatic drainage rom the head into the jugular vein leads to an accumulation o uid in jugular lymphatic sacs. Te birth prevalence o cystic hygromas approximates 1 in 5000. However, reecting the high in-utero lethality o the condition, the rst-trimester incidence exceeds 1 in 300 (Malone, 2005). Up to 70 percent o cystic hygromas are associated with aneuploidy. When cystic hygromas are diagnosed in the rst trimester, trisomy 21 is the most common aneuploidy, ollowed by 45,X and trisomy 18 (Kharrat, 2006; Malone, 2005).
First-trimester etuses with cystic hygromas are ve times more likely to be aneuploid than etuses with a thickened nuchal translucency. When cystic hygromas are diagnosed in the second trimester, approximately 75 percent o aneuploid cases are 45,X—urner syndrome (Johnson, 1993; Shulman, 1992). Even in the absence o aneuploidy, cystic hygromas coner a signicantly greater risk or other abnormalities, particularly ow-related cardiac deects. Tese include hypoplastic let heart and coarctation o the aorta (p. 292). Cystic hygromas may
FIGURE 15-26 Cleft lip/palate. A. This fetus has a prominent unilateral (left-sided) cleft lip. B. Transverse view of the palate in the same fetus demonstrates a defect in the alveolar ridge (arrow). The tongue (T) is also visible.
FIGURE 15-27 Micrognathia. A. Sagittal image of a fetus with severe micrognathia. B. 3-dimensional ultrasound rendering depicts the
recessed chin and downslanting palpebral fissures. C. A transverse image of the mandible was used to calculate a jaw index for this fetus. also be part o a genetic syndrome such as Noonan syndrome, an autosomal dominant disorder that shares several eatures with 45,X. Noonan syndrome is characterized by short stature, lymphedema, high-arched palate, and oten pulmonary valve stenosis.
Large cystic hygromas are usually associated with hydrops etalis, rarely resolve, and carry a poor prognosis. Small hygromas may undergo spontaneous resolution, and provided that etal karyotype and echocardiography results are normal, the prognosis may be good. Te likelihood o a nonanomalous liveborn neonate with normal karyotype ollowing identication o rst-trimester hygroma approximates 1 in 6 (Kharrat, 2006; Malone, 2005).
THORAX
Toracic anatomy imaged in the detailed anatomic survey may include the lungs, ribs, and diaphragm. Te lungs should appear homogeneous and symmetric, each occupying approximately one third o the area in the our-chamber view o the heart (Fig. 15-31). Te thoracic circumerence is measured at the skin line in a transverse plane at the level o the our-chamber view. I pulmonary hypoplasia is suspected secondary to a small thorax, such as with a severe skeletal dysplasia, comparison with a reerence table may be helpul (Appendix, p. 1238). Representative images o the diaphragm are usually obtained in the parasagittal or coronal plane. Te diaphragm appears as a hypoechoic line between the lungs and liver. Toracic abnormalities may appear sonographically as cystic or solid space-occupying lesions or as an eusion outlining the
FIGURE 15-28 Agnathia-otocephaly, ultrasound (A) and postdelivery (B) images. With this rare, lethal anomaly the mandible fails to develop, and the ears are inferiorly displaced and may be fused in the midline.
FIGURE 15-29 Epignathus, ultrasound (A) and postdelivery (B) images. This teratoma arises from the oral cavity or pharynx and may grow outward from the mouth or both outward and into the brain, as in this fetus (arrowhead). Arrows depict the external extent of the mass. (Reproduced with permission from Halima Abdirahman, RDMS.)
FIGURE 15-30 Cystic hygromas. A. This 9-week fetus with a cystic hygroma (arrow) was later found to have Noonan syndrome. B. Massive multiseptated hygromas (arrowheads) in the setting of hydrops fetalis at 15 weeks’ gestation. include the stomach bubble or bowel peristalsis in the chest and a wedgeshaped mass—the liver—located anteriorly in the let hemithorax.
Liver herniation complicates at least 50 percent o cases and is associated with a 30-percent reduction in the survival rate (Mullassery, 2010). With large lesions, impaired swallowing and mediastinal shit may result in hydramnios and hydrops, respectively.
An eort to reduce neonatal mortality rates and need or extracorporeal membrane oxygenation (ECMO) has ocused on prognostic indicators such as the sonographic lung-tohead ratio, described in Chapter 19 (p. 375). MR imaging parameters include measurements o lung volume and degree o liver herniation (Dutemeyer, 2020; Oluyomi-Obi, 2017; Worley, 2009).
■ Congenital Cystic Adenomatoid Malformation
Tis abnormality represents a hamartomatous overgrowth o terminal bronchioles that communicates with the t racheobronchial tree. It is also called congenital pulmonary airway malormation (CPAM), based on an understanding that not all histopathological types are cystic or adenomatoid (Azizkhan, 2008; Stocker, 1977 2002). Te estimated prevalence is 1 in 6000 to 8000 births, and this rate is rising because o improved sonographic detection o milder cases (Burge, 2010; Duncombe, 2002; Lau, 2017).
With ultrasound, a congenital cystic adenomatoid mal- ormation (CCAM) is as a well-circumscribed mass that may appear solid and echogenic or may have one or multiple variably sized cysts (Fig. 15-33). It usually involves one lobe, receives its blood supply rom the pulmonary artery, and has pulmonary venous drainage. Lesions with cysts ≥5 mm are generally termed macrocystic, and lesions that appear solid or have cysts <5 mm are microcystic (Adzick, 1985). In a review o 645 CCAM cases, the neonatal survival rate exceeded 95 percent, and 30 percent o cases demonstrated apparent prenatal resolution. Te other 5 percent o cases— typically very large lesions with associated mediastinal shit—were complicated by hydrops and had poor prognosis (Cavoretto, 2008). Microcystic CCAMs usually become less conspicuous with advancing gestation, because in addition to occupying less o the thorax, their echogenicity more closely resembles surrounding lung tissue. However, a subset o CCAMs may demonstrate rapid growth between 18 and 26 weeks’ gestation. Corticosteroid therapy has been used or large microcystic lesions to orestall growth and potentially ameliorate hydrops (Curran, 2010; Peranteau, 2016). I a large dominant cyst is present, thoracoamnionic shunt placement may lead to hydrops resolution. Fetal therapy or CCAM is discussed in Chapter 19 (p. 370). heart or lung(s). Fetal therapy or thoracic abnormalities is discussed in Chapter 19 (p. 376).
■ Diaphragmatic Hernia
Tis is a deect in the diaphragm through which abdominal organs herniate into the thorax. It is let-sided in approximately 75 percent o cases, right-sided in 20 percent, and bilateral in 5 percent (Gallot, 2007). Te prevalence o congenital diaphragmatic hernia (CDH) is 1 in 3000 to 5000 births (Cragan, 2009; Dolk, 2010). Associated anomalies and aneuploidy are ound in 40 percent o cases (Gallot, 2007; Stege, 2003). In population-based series, an associated abnormality reduces the overall survival rate o neonates with CDH rom approximately 50 percent to about 20 percent (Colvin, 2005; Gallot, 2007). Without other abnormalities, the major causes o neonatal mortality are pulmonary hypoplasia and pulmonary hypertension. Let-sided CDH typically causes dextroposition o the heart to the right side o the thorax, such that the cardiac axis points toward the midline (Fig. 15-32). Accompanying ndings
FIGURE 15-31 Normal thoracic anatomy. A. The lungs each occupy one third of the area in the four-chamber view of the heart. B. The diaphragm (arrow) appears as a hypoechoic line in between the lung and liver in this parasagittal view. LA = left atrium; LV = left ventricle; RA = right atrium; RV = right ventricle.
FIGURE 15-32 Congenital diaphragmatic hernia. In this transverse view of the thorax, the heart is shifted to the right side of the chest by a left-sided diaphragmatic hernia containing stomach (S), liver (L), and bowel (B).
■ Pulmonary Sequestration
Also called a bronchopulmonary sequestration, this abnormality is an accessory lung bud “sequestered” rom the tracheobronchial tree. It is nonunctioning lung tissue. Most cases diagnosed prenatally are extralobar, which means they are enveloped in their own pleura. Overall, however, most sequestrations present in adulthood and are intralobar—within the pleura o another lobe. Extralobar pulmonary sequestration is considered signi- cantly less common than CCAM, and no precise prevalence has been reported. Lesions have a let-sided predominance and most oten involve the let lower lobe. Associated anomalies have been reported in approximately 10 percent o cases (Yildirim, 2008).
Sonographically, pulmonary sequestration presents as a homogeneous, echogenic thoracic mass (Fig. 15-34A). Tus, it may resemble a microcystic CCAM. However, the blood supply is rom the systemic circulation—rom the aorta—rather than the pulmonary artery (Fig. 15-34B). Approximately 10 to 20 percent are located below the diaphragm. A small percentage o etuses with pulmonary sequestration develop a large ipsilateral pleural eusion, and without treatment, this may result in pulmonary hypoplasia or hydrops (Fig. 15-34C). reatment with thoracoamnionic shunting is discussed in Chapter 19 (p. 376). Hydrops may also result rom mediastinal shit or highoutput cardiac ailure due to the let-to-right shunt imposed by the mass. In the absence o a pleural eusion, the reported survival rate exceeds 95 percent, and 40 percent o cases demonstrate apparent prenatal resolution (Cavoretto, 2008).
■ Congenital High Airway Obstruction
Sequence
Tis rare anomaly usually results rom laryngeal or tracheal atresia. Te normal egress o lung uid is obstructed, and the tracheobronchial tree and lungs become massively distended. Sonographically, the lungs are brightly echogenic, the bronchi are dilated, the diaphragm is attened or everted, and the heart is compressed (Fig. 15-35). Impaired venous return leads to development o ascites, typically ollowed by hydrops. In one review o 118 cases, associated anomalies were identied in more than 50 percent (Sanord, 2012). Congenital high airway obstruction sequence (CHAOS) is a eature o the autosomal recessive Fraser syndrome and has been associated with the 22q11.2 deletion syndrome. In some cases, the obstructed airway spontaneously perorates, which potentially coners a better prognosis. Te EXI procedure has signicantly improved outcome in selected cases.
HEART
Cardiac malormations are the most common class o congenital anomalies. Teir overall prevalence is 8 cases in 1000 births (Cragan, 2009). Almost 90 percent o cardiac deects are multiactorial or polygenic in origin, another 1 to 2 percent result rom a single-gene disorder or gene-deletion syndrome, and 1 to 2 percent may occur rom exposure to a teratogen such as maternal diabetes or isotretinoin. Based on data rom population-based registries, approximately 1 in 8 liveborn and stillborn neonates with a congenital heart deect has a chromosomal abnormality (Dolk, 2010; Hartman, 2011). risomy 21 accounts or most o these cases, ollowed by trisomy 18, 22q11.2 deletion, trisomy 13, and monosomy X (Hartman, 2011). Approximately 50 to 70 percent o aneuploid etuses with cardiac anomalies are also ound to have noncardiac abnormalities. raditionally, congenital cardiac anomalies have been more challenging to detect than anomalies o other organ systems.
Recent series suggest that standard ultrasound may identiy 50 to 60 percent o those with major cardiac anomalies beore 22 weeks’ gestation (Byrne, 2020; Sun, 2018). Prenatal detection may improve neonatal survival, particularly or ductaldependent lesions, that is, those requiring prostaglandin inusion ater birth to keep the ductus arteriosus open (Franklin, 2002; Mahle, 2001; woretzky, 2001).
■ Standard Cardiac Examination
Standard cardiac assessment includes a our-chamber view, evaluation o rate and rhythm, evaluation o the let and right ventricular outow tracts, and when easible, documentation o the 3-vessel view and 3-vessel trachea view (Figs. 15-36 and
FIGURE 15-33 Congenital cystic adenomatoid malformation (CCAM). A. This left-sided microcystic CCAM is an echogenic mass that fills
the left hemithorax and causes mediastinal shift, which displaces the heart (arrows) to the right side of the chest. B. This left-sided macrocystic CCAM contains a cyst as large as the heart and also displaces the heart to the right. C. This right-sided CCAM contains multiple cysts of varying size (*) and displaces the heart farther to the left side of the chest (arrows).
FIGURE 15-34 Pulmonary sequestration. A. Transverse image of the thorax depicts a left lower lobe pulmonary sequestration (PS) in this 25-week fetus. B. Sagittal image showing that blood supply to the mass is from a branch of the abdominal aorta, which confirms the diagnosis. C. Over the next 3 weeks, a large ipsilateral pleural effusion develops (asterisk), resulting in mediastinal shift and dextroposition of the heart to the far-right thorax. D. After placement of a double-pigtail shunt through the chest wall, which drains the effusion into the amnionic fluid, the lung significantly reexpanded. Arrows point to coils of the pigtail shunt. (Reproduced with permission from Dr. Elaine Duryea.)
FIGURE 15-35 Congenital high airway obstruction sequence (CHAOS). The lungs (L) appear brightly echogenic, and the bronchi (arrow) are dilated with fluid. Flattening and eversion of the diaphragm is common, as is ascites (asterisks).
FIGURE 15-36 Diagram showing measurement of cardiac axis from the four-chamber view. LA = left atrium; LV = left ventricle; RA = right atrium; RV = right ventricle. 15-37). It is hoped that examination o the cardiac outow tracts and 3-vessel views will improve detection o outow tract abnormalities that may have a normal-appearing our-chamber view, such as tetralogy o Fallot and transposition o the great arteries. Centers with expertise have reported at least 90 percent detection o such anomalies with incorporation o these views (Brandt, 2015; Palatnik, 2016).
Te our-chamber view is a transverse image o the etal thorax at a level immediately above the diaphragm. It allows evaluation o cardiac size, position in the thorax, cardiac axis, atria and ventricles, oramen ovale, atrial septum primum, interventricular septum, and atrioventricular valves (Fig. 15-37A).
Te let ventricle is apex-orming, and two pulmonary veins are oten visible entering the let atrium. Te atria and ventricles should be similar in size, and the apex o the heart should orm a 45-degree angle with the let anterior chest wall (see Fig. 15-36). Abnormalities o cardiac axis are encountered in more than one third o etuses with structural cardiac anomalies (Shipp, 1995; Crane, 1997; Sinkovskaya, 2015).
Te let ventricular outfow tract view demonstrates that the ascending aorta arises entirely rom the let ventricle. Te interventricular septum is shown to be in continuity with the anterior wall o the aorta, and the mitral valve in continuity with the posterior wall o the aorta (Fig. 15-37B). Ventricular septal
A B C
D E F
G H I
PA
DA
AA
T
SVC
Ao
SVC
FIGURE 15-37 Standard and detailed examination cardiac views. A. Four-chamber view. B. Left ventricular outflow tract view. Arrow depicts mitral valve becoming the posterior wall of the aorta. The arrow with asterisk marks the interventricular septum becoming the anterior aortic wall. C. Right ventricular outflow tract view. D. Three-vessel view. E. Three-vessel trachea view F. High short-axis view (outflow tracts). G. Aortic arch view. H. Ductal arch view. I. Superior and inferior vena cavae views. AA = aortic arch; Ao = aorta; DA = ductal arch; IVC = inferior vena cava; LA = left atrium; LV = left ventricle; PA = pulmonary artery; RA = right atrium; RV = right ventricle; SVC = superior vena cava; T = trachea.
deects and outow tract abnormalities are oten visible in this view. Te right ventricular outfow tract view shows the right ventricle giving rise to the main pulmonary artery, which subsequently branches into the right and let pulmonary arteries. (Fig. 15-37C,F). ogether, the let and right outow tract views demonstrate the normal perpendicular orientation o the aorta and pulmonary artery and the comparable size o these great arteries.
Te 3-vessel view (3VV) is a transverse image obtained just above the base o the heart. Te three vessels line up in a row: an oblique view o the pulmonary artery, which should appear long and cylindrical; a cross-sectional image o the ascending aorta; and a cross-sectional image o the superior vena cava (SVC) (Fig. 15-37D). Te pulmonary artery and aorta should be similar in diameter. Te 3-vessel trachea view (3VV) also is a transverse image but is obtained urther cephalad. It includes the pulmonary artery giving rise to the ductus arteriosus as it makes a V-shape with the aortic arch, along with the SVC and the trachea (Fig. 15-37E). Te 3VV can be helpul or identiying aortic arch abnormalities, particularly ductal-dependent lesions.
■ Specialized Cardiac Examination
Te detailed ultrasound examination includes the ve cardiac components o the standard examination plus the superior and inerior vena cavae and the aortic and ductal arch views (Fig. 15-37G–I). Te examination also involves evaluation o the interventricular septum and cardiac situs, documentation o which may be acilitated with video clips. Fetal echocardiography is a more extensive examination o cardiac structure and unction designed to characterize abnormalities. In addition to grayscale imaging views, echocardiography includes evaluation o cardiac rate and rhythm, color Doppler ultrasound, pulsed-wave Doppler ultrasound, cardiac biometry, and i clinically relevant, cardiac unction assessment (American Institute o Ultrasound in Medicine, 2020b). Tese components are beyond the scope o this text. Echocardiography indications are discussed in Chapter 14 (p. 252). Selected cardiac anomalies are reviewed next.
Ventricular Septal Defect
Tis is the most common congenital cardiac anomaly. It is ound in approximately 1 in 300 births (Bjornard, 2013; Cragan, 2009; Dolk, 2010). A deect may be appreciated in the perimembranous or muscular portion o the interventricular septum in the our-chamber view, and imaging o the let ventricular outow tract may show discontinuity o the interventricular septum as it becomes the wall o the aorta (Fig. 15-38). Color Doppler may demonstrate ow across the deect. Genetic abnormalities are diagnosed with chromosomal microarray analysis in approximately 1 percent o isolated cases but ound in at least 15 percent i other structural abnormalities also are present (Maya, 2020). More than a third o prenatally diagnosed ventricular septal deects close in utero, and another third close in the rst year o lie (Cho, 2017; Paladini, 2002; Svirsky, 2019).
Endocardial Cushion Defect
Tis is also called an atrioventricular (AV) septal deect or AV canal deect. It has a birth prevalence o approximately 1 in 2500 and is associated with trisomy 21 in more than hal o cases (Christensen, 2013; Cragan, 2009; Dolk, 2010). Te endocardial cushions are the crux o the heart, and deects jointly involve the atrial septum primum, interventricular septum, and medial leaets o the mitral and tricuspid valves (Fig. 15-39).
A B
FIGURE 15-38 Ventricular septal defect. A. In this four-chamber view, a defect (arrow) is noted in the perimembranous portion of the interventricular septum. B. The left ventricular outflow tract view demonstrates a break in continuity (arrow) between the interventricular septum and the anterior wall of the aorta.
A B
FIGURE 15-39 Endocardial cushion defect. A. During ventricular systole, the lateral leaflets of the mitral and tricuspid valves come together in the midline. But the atrioventricular valve plane is abnormal, a common atrium (A) is observed, and there is a visible defect (arrow) in the interventricular septum. B. During diastolic filling, opening of the atrioventricular valves more clearly demonstrates the absence of their medial leaflets.
Te majority o deects are balanced–with ventricles comparable in size. Some are unbalanced however, with one side o the heart signicantly smaller than the other. Te endocardial cushion deect is considered partial i there is absence o the atrial septum primum and a more subtle AV valve plane abnormality, with no ventricular septal deect.
Endocardial cushion deects are a common nding in heterotaxy, also known as cardiosplenic or isomerism syndromes. Heterotaxy implies that thoraco-abdominal organs normally on one side o the body are on an incorrect side, on both sides, or absent, as in polysplenia or asplenia. Complex cardiac abnormalities are a common eature, particularly endocardial cushion deects. In a review o 632 pregnancies with etal heterotaxy, an endocardial cushion deect was present in 60 percent (Buca, 2018). Tese endocardial cushion deects are a particularly atrisk group because o their association with third-degree heart block, which coners a poor prognosis.
Hypoplastic Left Heart Syndrome
Tis anomaly is ound in approximately 1 in 4000 births (Bjornard, 2013; Cragan, 2009; Dolk, 2010;). During secondtrimester ultrasound examination, the let ventricle may be so small that a chamber is difcult to appreciate (Fig. 15-40). Alternately, the let ventricle may be normal in size or dilated but have severely decreased contractility and an echogenic inner wall due to endocardial broelastosis. Tere may be no visible let ventricular inow or outow, and reversal o ow may be documented in the aortic arch in the 3-vessel trachea view. As gestation advances, the let ventricle and aorta become progressively smaller. Prenatal detection is nearly 90 percent in population-based registries.
Hypoplastic let heart syndrome is a ductal-dependent lesion or which neonatal administration o prostaglandin therapy is essential. Postnatal treatment consists o a three-staged palliative repair—single ventricle palliation, or cardiac transplantation. Rates o survival to adulthood may reach 70 percent (Feinstein, 2012). However, morbidity is high, and developmental delays are common (Lloyd, 2017; Paladini, 2017; Wilson, 2018). Fetal therapy or this condition is discussed in Chapter 19 (p. 378).
Coarctation of the Aorta
Coarctation reers to narrowing o the aortic arch. Te birth prevalence approximates 1 in 2500 (Bjornard, 2013). Usually, the narrowing is ocal and ound just distal to the origin o the let subclavian artery at the aortic isthmus. Alternatively, a long segment o the aorta may be aected. Coarctation may be isolated or associated with other cardiac anomalies, such as hypoplastic let heart syndrome, unbalanced endocardial cushion deect, or double-outlet right ventricle. Te most common sonographic nding is disproportion in ventricular size, with the let ventricle smaller than the right. However, only one third with this nding have coarctation (Ghi, 2018; van Nisselrooij, 2018). Other ndings include narrowing o the aortic arch in the 3-vessel trachea view or narrowing o the isthmus in the aortic arch view–which may be challenging to image. Coarctation is associated with urner syndrome (45,X), with the 22q11.2 deletion, and with autosomal trisomies.
Ebstein Anomaly
Tis rare anomaly is characterized by apical displacement o the tricuspid valve, such that the septal and posterior valve leaets o the tricuspid valve attach closer to the cardiac apex (Fig. 15-41). Te birth prevalence is approximately 1 in 20,000 (Boyle, 2017). Fetuses with Ebstein anomaly develop varying degrees o tricuspid regurgitation. In severe cases, right atrium becomes markedly dilated, and the etus may develop cardiomegaly and hydrops. For many years, there was concern that lithium exposure predisposed to Ebstein anomaly. However, as discussed in Chapter 8 (p. 154), the absolute attributable risk is likely well below 1 percent.
Tetralogy of Fallot
Tis anomaly occurs in approximately 1 in 3000 births (Cragan, 2009; Dolk, 2010; Nelson, 2016). It includes a ventricular septal deect; an overriding aorta; a pulmonary valve or pulmonary artery abnormality, and right ventricular hypertrophy (Fig. 15-42). Te last does not usually maniest beore birth. Due to the location o the ventricular septal deect, the ourchamber view may appear normal.
Aortic_Arch
FIGURE 15-40 Hypoplastic left heart syndrome. A. In this four-chamber view, the left ventricle appears “filled in” and echogenic, due to endocardial fibroelastosis. B. Color Doppler depicts only flow from the right atrium into the right ventricle, and no left ventricular filling is visible. C. Color Doppler shows reversal of flow in the aortic arch (arrow), which is perfused retrograde via the ductus arteriosus. LA = left atrium; LV = left ventricle; RA = right atrium; RV = right ventricle.
Te pulmonary artery abnormality is pulmonary stenosis in 60 percent o cases, pulmonary atresia in slightly more than 25 percent, and absent pulmonary valve in 10 to 15 percent (Zhao, 2016). Absence o the pulmonary valve leads to marked enlargement o the pulmonary artery and poses a risk or hydrops. Te enlarged pulmonary artery can also compress the trachea and cause tracheomalacia.
Chromosomal abnormalities are identied in approximately one third o etuses with tetralogy o Fallot. O these, 22q11.2 deletions compose 15 to 20 percent, and autosomal trisomies constitute 10 percent (Zhao, 2016). D-Transposition of the Great Arteries Tis anomaly is characterized by outow tracts that arise in parallel rom the ventricles. Te right ventricle gives rise to the aorta, and the let ventricle to the pulmonary artery (Fig. 15-43). Tus, there is ventriculo-arterial discordance. Te birth prevalence approximates 1 in 4000 (Bjornard, 2013). Te our-chamber view is oten normal. Prenatal detection approximates 40 percent but is thought to improve with visualization o the outow tracts (Ravi, 2018).
Additionally, the 3-vessel views may demonstrate only two vessels, because the pulmonary artery is underneath the aorta.
D-transposition contrasts with
L- or corrected transposition o the great arteries, in which there is atrioventricular discordance in addition to ventricular-arterial discordance.
L-transposition is strongly associated with other cardiac anomalies and is much less likely to be diagnosed prenatally as an isolated nding.
Double-outlet Right Ventricle
With this anomaly, the majority o blood ow to both the pulmonary artery and the aorta comes rom the right ventricle. Double-outlet right ventricle (DORV) is always associated with a ventricular septal deect. DORV has a spectrum o presentation. It is categorized according to the location o the ventricular septal deect and the relative proportion o blood ow rom the right ventricle to the outow tracts. Te outow tracts are oten malposed, arising in parallel. Te birth prevalence approximates 1 in 20,000 (Bjornard, 2013).
Truncus Arteriosus
Tis rare anomaly is characterized by a single, large outow tract exiting the heart—a common arterial trunk—rather than a separate aorta and pulmonary artery. A prominent ventricular septal deect is usually identied, with an enlarged overriding outow tract that gives rise to pulmonary arteries as well as head and neck vessels (Fig. 15-44). Te dierential diagnosis includes tetralogy o Fallot with pulmonary atresia. Te birth prevalence o truncus arteriosus approximates 1 in 16,000 (Bjornard, 2013). In 1949, Collett and Edwards categorized our types o truncus arteriosus according to the origin o the
A
LV
LA
RV
RA
B
FIGURE 15-41 Ebstein anomaly. A. In this four-chamber view, the tricuspid valve’s septal leaflet attaches closer to the cardiac apex (arrowhead) than the corresponding mitral valve leaflet (arrow). B. The color Doppler blue jet reflects tricuspid regurgitation. LA = left atrium; LV = left ventricle; RA = right atrium; RV = right ventricle.
A B C
FIGURE 15-42 Tetralogy of Fallot. A. Left ventricular outflow tract view shows a ventricular septal defect with an overriding aorta. The arrow points to the aortic valve. B. Right ventricular outflow tract view demonstrating severe pulmonary artery stenosis. The arrow points to the pulmonary valve. C. In this fetus with tetralogy of Fallot with absent pulmonary valve, the pulmonary artery shows marked enlargement.
Ao = aorta; LV = left ventricle; PA = pulmonary artery; RV = right ventricle.
FIGURE 15-43 D-transposition of the great arteries. A. Transverse image with color Doppler depicting the outflow tracts arising in parallel.
B. View of the left ventricle giving rise to the pulmonary artery, which subsequently branches (arrows). C. Sagittal image of the aorta arising anteriorly from the right ventricle and parallel to the pulmonary artery, which arises posteriorly. Ao = aorta; LV = left ventricle; PA = pulmonary artery; RV = right ventricle.
FIGURE 15-45 Rhabdomyoma. In this four-chamber view of the heart, a large, echogenic, well-circumscribed mass (R) fills the right ventricle and abuts the tricuspid valve (arrow). Despite its size, the mass did not obstruct flow, and the neonate did well.
A B
FIGURE 15-44 Truncus arteriosus. Grayscale (A) and color Doppler (B) images depict a single, large outflow tract. The common arterial trunk (CAT) overlies a ventricular septal defect (arrowhead) and gives rise to the head and neck vessels and pulmonary arteries.
LV = left ventricle; RV = right ventricle. (Reproduced with permission from Paul Mallamaci, RDMS.)
pulmonary arteries. Characterization o anatomic variants can oten only be made postnatally.
Cardiac Rhabdomyoma
Tis is the most common cardiac tumor. Approximately 50 percent o cases are associated with tuberous sclerosis, an autosomal dominant disease with multiorgan system maniestations. uberous sclerosis is caused by mutations in the hamartin (TSC1) and tuberin (TSC2) genes.
Sonographically, cardiac rhabdomyomas are well-circumscribed echogenic masses, usually within the ventricles or out- ow tracts (Fig. 15-45). Tey may be single or multiple, may grow in size during gestation, and may occasionally lead to inow or outow obstruction. In cases without obstruction or large tumor size, the prognosis is relatively good rom a cardiac standpoint, because the tumors tend to regress ater the neonatal period. Because extracardiac ndings o tuberous sclerosis may not be apparent with prenatal sonography, MR imaging may be considered to evaluate etal CNS anatomy (p. 266).
■ MMode
Motion-mode or M-mode imaging is a linear display o cardiac cycle events, with time on the x-axis and motion on the y-axis. It is oten used to measure embryonic or etal heart rate (Fig. 14-1, p. 248). I an abnormality o heart rate or rhythm is identied, M-mode imaging permits separate evaluation o atrial and ventricular waveorms. Tus, it is particularly useul or characterizing arrhythmias and their response to treatment (Chap. 19, p. 367). M-mode can also be used to assess ventricular unction and atrial and ventricular outputs.
Premature Atrial Contractions
Also called atrial extrasystoles, these are the most common etal arrhythmia and a requent nding. Tey represent cardiac conduction system immaturity and oten resolve with advancing gestation or in the neonatal period. Premature atrial contractions (PACs) may be conducted and thus sound like extra beats. However, they are more commonly blocked, and with handheld Doppler they sound like dropped beats. As shown in Figure 15-46, the dropped beat may be demonstrated with M-mode evaluation as a compensatory pause that ollows the premature contraction.
PACs are not associated with major structural cardiac abnormalities. Older case reports describe an association with maternal caeine consumption and with hydralazine (Lodeiro, 1989; Oei, 1989). In approximately 2 percent o cases, aected etuses are later identied to have supraventricular tachycardia (SVT), which is an arrhythmia that requires urgent treatment (Copel, 2000). Accordingly, pregnancies with etal PACs are oten ollowed with etal heart rate assessment as oten as every 1 to 2 weeks until the ectopy resolves. reatment o etal SV and other arrhythmias is discussed in Chapter 19 (p. 368).
ABDOMEN
Abdominal anatomy visible in the second and third trimester includes the stomach, liver, gallbladder, spleen, adrenal glands, kidneys, renal arteries, small and large bowel, and ventral wall. Te stomach is nearly always identied ater 14 weeks’ gestation. Nonvisualization o the stomach may be secondary to impaired swallowing in the setting o oligohydramnios or to underlying causes such as esophageal atresia, a cranioacial anomaly, or a CNS or musculoskeletal abnormality. Fetuses with hydrops can also have impaired swallowing.
Both the liver and spleen may be viewed in a transverse image obtained at the level o the stomach and intrahepatic portion o the umbilical vein—the plane at which the abdominal circumerence is measured (see Fig. 15-2B). Hepatosplenomegaly may occur with congenital inection or with hydrops. By convention, the liver is measured in the sagittal or coronal plane, rom the top o the right hemidiaphragm to the inerior tip o the right lobe (Fig. 15-47). Te spleen is posterior to the stomach in the transverse plane. Te gallbladder may be imaged just inerior to the level at which the abdominal circumerence is measured. It lies to the right o the intrahepatic portion o the umbilical vein and has a conical or teardrop shape (see Fig. 15-47B).
Te appearance o etal bowel changes with maturation. Increased bowel echogenicity may indicate a small amount o swallowed intraamnionic blood, especially i the maternal serum alpha-etoprotein level is elevated. Te bowel appears as bright as bone in approximately 0.5 percent o second-trimester etuses.
In such cases, the risk or etal trisomy 21 is increased (Fig. 17-3, p. 340). Echogenic bowel is also associated with etal cytomegalovirus inection and with cystic brosis, in which echogenicity represents inspissated meconium.
FIGURE 15-46 M-mode. This image demonstrates normal concordance between atrial (A) and ventricular (V) contractions. Movement of the tricuspid valve (T) is also shown. The blue arrow denotes a premature atrial contraction followed by a compensatory pause.
FIGURE 15-47 Abdominal organs. A. The liver is measured from the top of the right hemidiaphragm to the inferior tip of the right lobe in this coronal image. B. Transverse image of the abdomen just inferior to the level at which the abdominal circumference is measured, depicting the gallbladder (G), stomach (S), spleen (Sp), liver (L), and right adrenal gland (A).
■ Gastrointestinal Obstruction
Bowel atresia is characterized by obstruction and proximal bowel dilation. In general, the more proximal the obstruction, the more likely it is to lead to hydramnios. Te degree o hydramnios rom proximal small-bowel obstruction can be sufciently severe to result in maternal respiratory compromise or preterm labor and may necessitate amnioreduction (Chap. 14, p. 258).
Esophageal Atresia
Te birth prevalence o this anomaly approximates 1 in 4000 (Cragan, 2009; Pedersen, 2012). It may be suspected when there is no visible stomach bubble or when the stomach is contracted. However, because esophageal atresia is associated with a tracheoesophageal stula in up to 90 percent o cases, uid is oten able to enter the stomach. More than 50 percent o those with esophageal atresia have other abnormalities or underlying genetic syndromes. Multiple mal- ormations are present in 30 percent o cases, and aneuploidy such as trisomy 18 or 21, in 10 percent.
Approximately 10 percent o cases o esophageal atresia is ound as part o the VACERL association (Pedersen, 2012).
Duodenal Atresia
Tis anomaly occurs in approximately 1 in 10,000 births (Best, 2012; Dolk, 2010). It is characterized by the sonographic doublebubble sign, which develops rom distention o the stomach and the rst part o the duodenum (Fig. 15-48). Tis nding may not be obvious beore 24 weeks’ gestation. Demonstrating continuity between the stomach and proximal duodenum conrms that the second “bubble” is the proximal duodenum. Approximately 30 percent o aected etuses have an associated chromosomal abnormality or genetic syndrome, particularly trisomy 21. O cases without a genetic abnormality, a third have associated anomalies, most commonly cardiac deects and other gastrointestinal abnormalities (Best, 2012).
Jejunoileal Atresia
Tis condition may present with dilated small-bowel loops that ll the abdomen or with meconium peritonitis rom bowel per- oration (Fig. 15-49). Associated gastrointestinal abnormalities are identied postnatally in 25 percent o cases, with malrotation in 10 to 15 percent (Stollman, 2009). Cystic brosis also is identied in approximately 10 percent. In general, jejunal atresia is more strongly associated with bowel dilation and hyperperistalsis, and ileal atresia, with per- oration. Bowel dilatation in jejunal atresia typically does not present until ater 24 weeks’ gestation and may be accompanied by hydramnios. Peroration is requently associated with ascites, and bright echoes may be visible outside the bowel lumen, outlining the peritoneal cavity. Over time, the ascites resolves, and the extravasated meconium may orm a pseudocyst. Fetal MR imaging can assist with identiying the level o the deect (Chap. 14, p. 267).
Anal Atresia
Also known as imperorate anus, this condition is less readily diagnosed by sonography. Hydramnios is not a typical eature, and the bowel may not be signicantly dilated. A transverse view through the pelvis may show an enlarged rectum as an anechoic structure between the bladder and the sacrum. Anal atresia is a eature o the VACERL association.
FIGURE 15-48 Duodenal atresia. Transverse image of the double-bubble sign, which represents distension of the stomach (S) and the first part of the duodenum (D). Continuity between these structures confirms that the second cystic structure is the duodenum.
FIGURE 15-49 Jejunoileal atresia with meconium peritonitis. A. Sagittal image of the fetal abdomen at 26 weeks’ gestation with severe ascites following bowel perforation. Arrows point to the bowel, which is considerably more echogenic than the liver. B. Transverse image at 30 weeks demonstrates resolution of ascites. The abdomen is now filled with dilated loops of small bowel. Focal bright echoes (arrows) represent extraluminal meconium. A = ascites; L = liver.
■ Ventral Wall Defects
Te integrity o the abdominal wall is assessed at the level o the cord insertion during the standard examination (Fig. 15-50). Ventral wall deects include gastroschisis, omphalocele, and body stalk anomaly.
Gastroschisis
Tis is a ull-thickness abdominal wall deect located to the right o the umbilical cord insertion. Bowel herniates through the deect into the amnionic cavity. Gastroschisis may be diagnosed as early as the late rst trimester (Fig. 15-51). Te prevalence is approximately 1 in 2000 births (Jones, 2016; Nelson, 2015). Gastroschisis is the one major anomaly more common in etuses o younger mothers, and the average maternal age is 20 years (Santiago-Muñoz, 2007). Coexisting bowel abnormalities such as jejunal atresia are ound in approximately 15 percent o cases (Overcash, 2014). Gastroschisis is not associated with aneuploidy, and the survival rate is 90 to 95 percent (Nelson, 2015; Raitio, 2020).
Fetal-growth restriction complicates gastroschisis in 15 to 40 percent o cases (Overcash, 2014; SantiagoMuñoz, 2007). Growth restriction does not appear to coner increased mortality (Nelson, 2015; Overcash, 2014). However, earlier gestational age at delivery does pose a risk or adverse outcome, and planned delivery at 36 to 37 weeks’ gestation has not been ound to benet the neonate (Al-Ka, 2016; South, 2013).
Omphalocele
Te birth prevalence o this anomaly approximates 1 in 3000 to 5000 (Can- eld, 2006; Dolk, 2010). It develops when the lateral ectomesodermal olds ail to meet in the midline. Abdominal organs herniate into the base o the umbilical cord, covered only by a two-layered sac o amnion and peritoneum (Fig. 15-52). More than hal o cases are associated with other major anomalies or aneuploidy. Aneuploidy is particularly common with smaller deects (De Veciana, 1994). Omphalocele is also a component o several syndromes, including Beckwith–Wiedemann, cloacal exstrophy, and pentalogy o Cantrell. Neonatal survival approximates 90 percent or isolated cases and 80 percent in those with other structural abnormalities (Raitio, 2021; Springett, 2014). Isolated deects containing liver are typically delivered via cesarean to decrease the risk or etal trauma and bleeding.
Body Stalk Anomaly
Also known as limb-body-wall complex or cyllosoma, this rare, lethal anomaly is characterized by abnormal body wall ormation. ypically, no abdominal wall is visible, and the abdominal organs extrude into the extraamnionic coelom (Fig. 15-53). Te body and placenta are closely approximated or used, and
FIGURE 15-50 Normal ventral wall. Transverse view of the abdomen demonstrating normal umbilical cord insertion and integrity of the anterior abdominal wall.
A B
FIGURE 15-51 Gastroschisis. Transverse views of the lower abdomen at 13 weeks’ gestation (A) and 18 weeks (B) depict multiple small bowel loops (B) that have herniated into the amnionic cavity through a defect to the right of the cord insertion (arrow).
FIGURE 15-52 Omphalocele. Transverse view of the abdomen showing a large, round, membrane-covered ventral wall defect containing exteriorized liver.
the umbilical cord is extremely short. Acute-angle scoliosis is another eature, and amnionic bands are oten identied.
■ Kidneys and Genitourinary Tract
Te etal kidneys are visible adjacent to the spine, requently in the rst trimester and routinely by 18 weeks’ gestation (Fig. 15-54). Te length o the kidney approximates 20 mm at 20 weeks and grows about 1.1 mm each week thereater (Chitty, 2003).
Te etal bladder is also readily seen in the second trimester as a round, anechoic structure in the anterior midline o the pelvis. With application o Doppler, the bladder is outlined by the two superior vesical arteries as they become the umbilical arteries o the umbilical cord (Chap. 6, p. 114). Te etal ureters and urethra are not visible sonographically unless abnormally dilated. Ater 18 weeks’ gestation, the kidneys are the major source o amnionic uid (Chap. 14, p. 256). Normal amnionic uid volume in the second hal o pregnancy suggests urinary tract patency and at least one unctioning kidney.
External genitalia are a component o the detailed examination and are part o the standard examination in multietal gestations and when medically indicated (American Institute o Ultrasound in Medicine, 2019). Identication may aid counseling in pregnancies at risk or an X-linked genetic condition. Examples o normal and ambiguous genitalia are shown in
Figure 15-55. Disorders o sexual development are discussed in Chapter 3 (p. 35).
Renal Pelvis Dilatation
Present in 1 to 5 percent o etuses, this nding is also called urinary tract dilatation or hydronephrosis. In 40 to 90 percent o cases, and particularly when mild, renal pelvis dilatation is transient or physiological and does not represent an underlying abnormality (Ismaili, 2003; Nguyen, 2010). In approximately one third o cases, a urinary tract abnormality is conrmed in the neonatal period. O these, ureteropelvic junction (UPJ) obstruction and vesicoureteral refux (VUR) are the most requent. Te etal renal pelvis diameter is measured anterior-to-posterior in a transverse plane. Calipers are placed on the inner border o the uid collection (Fig. 15-56). Although various thresholds have been dened, the pelvis is typically considered dilated i it exceeds 4 mm in the second trimester or 7 mm at approximately 32 weeks’ gestation (Nguyen, 2014; Reddy, 2014). Te second-trimester threshold is used to identiy pregnancies that warrant subsequent third-trimester evaluation.
Te Society or Fetal Urology categorized renal pelvis dilatation based on a metaanalysis o more than 100,000 screened pregnancies (Table 15-2) (Lee, 2006; Nguyen, 2010). Provided that the nding is isolated, the degree o dilation correlates with
A B
FIGURE 15-53 Body-stalk anomaly. A. This 15-week fetus has no visible abdominal wall. The thorax is disproportionately small compared with the head, and the small lower torso is deviated to the side. Arrows point to the large mass of extruded abdominal organs. B. Acuteangle scoliosis. Arrows depict the abnormal curvature of the spine. (Reproduced with permission from Deirdre Snelson, RDMS.)
A B
FIGURE 15-54 Normal fetal kidneys and bladder. A. The kidneys are visible adjacent to the spine in this 29-week fetus. A small amount of urine is often visible within the renal pelvis (arrow). B. Normal fetal bladder, outlined by the two superior vesical arteries as they become the umbilical arteries.
dilatation. Te birth prevalence is 1 in 1000 to 2000, and males are aected three times more oten than emales (Williams, 2007; Woodward, 2002). Obstruction is generally unctional rather than anatomical, and it is bilateral in up to a ourth o cases. Te likelihood o UPJ obstruction ranges rom 5 percent with mild renal pelvis dilatation to more than 50 percent with severe dilatation (Lee, 2006).
Duplicated Renal Collecting System. In this anatomical anomaly, the upper and lower poles o the kidney—called moieties— are each drained by a separate ureter (Fig. 15-57). Duplication is ound in approximately 1 in 4000 pregnancies, is more common in emales, and is bilateral in 15 to 20 percent o cases (James, 1998; Vergani, 1998; Whitten, 2001). Sonographically, an intervening tissue band separates two distinct renal pelves. Hydronephrosis or ureteral dilation may develop due to abnormal implantation o one or both ureters within the bladder—a relationship that reects the anatomical Weigert-Meyer rule. Te upper pole ureter tends to develop obstruction rom a ureterocele within the bladder, whereas the lower pole ureter has a shortened intravesical segment that predisposes to VUR (see Fig. 15-57). Tus, both moieties may become dilated rom dierent etiologies, and both are at risk or loss o unction.
Renal Agenesis
Te estimated prevalence o bilateral renal agenesis is 1 in 8000 births, whereas that o unilateral renal agenesis is 1 in 1000 births (Cragan, 2009; Dolk, 2010; Sheih, 1989; Wiesel, 2005). With an absent kidney, color Doppler imaging o the descending aorta demonstrates absence o the ipsilateral renal artery (Fig. 15-58). In addition, the ipsilateral adrenal gland typically enlarges to ll the renal ossa, termed the lying down adrenal sign (Homan, 1992).
I renal agenesis is bilateral, no urine is produced, and the resulting anhydramnios leads to pulmonary hypoplasia, limb contractures, and distinctive acies. When this combination results rom renal agenesis, it is called Potter syndrome, ater Dr. Edith Potter, who described it in 1946. When these abnormalities result rom severely decreased amnionic uid volume rom another etiology, such as bilateral multicystic dysplastic kidney or autosomal
TABLE 15-2. Risk for Postnatal Urinary Abnormality
According to Degree of Renal Pelvis Dilation
Dilation
Second
Trimester Third Trimester
Postnatal
Abnormality
Mild 4 to <7 mm 7 to <9 mm 12%
Moderate 7 to ≤10 mm 9 to ≤15 mm 45%
Severe >10 mm >15 mm 88%
Modified from Lee, 2006; Nguyen, 2010.
A B
FIGURE 15-56 Renal pelvis dilatation. A. The anterior-posterior diameter of the renal pelves measured 7 mm in the transverse plane in this 34-week fetus. B. Sagittal image at 32 weeks depicts renal pelvis dilatation with rounded calyces (arrow) in the setting of ureteropelvic junction obstruction.
A B
C D
FIGURE 15-55 Normal and ambiguous genitalia. A. Female labia. B. Male penis and scrotum. C. Ambiguous genitalia with a phallus and bifid scrotum. D. With this condition, color Doppler can assist locating the urethral meatus, which is pinpointed here by the urine stream (blue).
the likelihood o an underlying abnormality. Associated calyceal dilation, cortical thinning, or dilation elsewhere along the urinary tract coner increased risk (Nguyen, 2014). Mild pyelectasis in the second trimester is a minor aneuploidy marker associated with a slightly increased risk or trisomy 21, particularly when other ndings or risk actors are also present (Fig. 17-3, p. 340). Ureteropelvic Junction Obstruction. Tis condition is the most common abnormality associated with renal pelvis recessive polycystic kidney disease, it is called Potter sequence. Te prognosis or these abnormalities is extremely poor.
Multicystic Dysplastic Kidney
Tis severe orm o renal dysplasia results in a nonunctioning kidney. Te nephrons and collecting ducts orm abnormally, such that primitive ducts are surrounded by bromuscular tissue, and the ureter is atretic (Hains, 2009). Sonographically, the kidney contains numerous, variably sized, smooth-walled cysts that do not communicate with the renal pelvis and are surrounded by echogenic cortex (Fig. 15-59). Unilateral multicystic dysplastic kidney (MCDK) has a prevalence o 1 in 4000 births. Contralateral renal abnormalities are present in 30 to 40 percent—most requently VUR or UPJ obstruction (Schreuder, 2009). Nonrenal anomalies have been reported in 25 percent o cases. Cystic dysplasia may occur as a component o many genetic syndromes (Lazebnik, 1999; Schreuder, 2009). I MCDK is isolated and unilateral, the prognosis is generally good. Bilateral MCDK is ound in approximately 1 in 12,000 births. It is associated with severely decreased amnionic uid volume starting early in gestation. Tis leads to Potter sequence and a poor prognosis.
Polycystic Kidney Disease
O the hereditary polycystic diseases, only the inantile orm o autosomal recessive polycystic kidney disease (ARPKD) may be reliably diagnosed prenatally. ARPKD is a chronic, progressive disease o the kidneys and liver that results in cystic dilation o the renal collecting ducts and in congenital hepatic brosis (urkbey, 2009). Te carrier requency o a disease-causing mutation in the PKHD1 gene approximates 1 in 70, and the birth prevalence is 1 in 20,000 (Zerres, 1998). Te phenotypes o ARPKD range rom lethal pulmonary hypoplasia at birth to a presentation in late childhood or even adulthood with predominantly hepatic maniestations. Sonographically, ARPKD displays abnormally large kidneys that may ll or even distend the etal abdomen and have a solid, ground-glass texture (Fig. 15-60). Severe oligohydramnios coners a poor prognosis.
Autosomal dominant polycystic kidney disease (ADPKD), which is ar more common, usually does not maniest until adulthood (Chap. 56, p. 1001). Even so, some etuses with ADPKD have mild renal enlargement and enhanced renal echogenicity in the setting o normal amnionic uid volume.
A B
FIGURE 15-57 Duplicated renal collecting system. A. Renal pelvis dilatation is visible in both the upper (U) and lower (L) pole moieties, which are separated by an intervening band of renal tissue (arrowhead). B. A ureterocele (arrowhead) is visible within the bladder. Color Doppler depicts the superior vesical arteries.
A B
FIGURE 15-58 Renal agenesis. A. Coronal image of a fetus with bilateral renal agenesis in which color Doppler of the abdominal aorta is used to demonstrate absence of the renal arteries. B. In this fetus with unilateral renal agenesis, arrowheads point to the adrenal gland (arrowheads) filling the renal fossa, which is the “lying-down” adrenal sign.
FIGURE 15-59 Multicystic dysplastic kidneys. Coronal view of the fetal abdomen demonstrates enlarged kidneys that are filled with cysts of varying size and contain no visible renal pelvis or normalappearing renal tissue.
Te dierential diagnosis or these ndings includes several genetic syndromes, aneuploidy, or normal variant.
Bladder Outlet Obstruction
Distal obstruction o the urinary tract is more requent in male etuses, and the most common etiology is posterior urethral valves. It may be diagnosed as early as the rst trimester in some cases (Fig. 15-61). Te bladder is markedly dilated, with accompanying dilation o the proximal urethra, which is termed the “keyhole” sign. Oligohydramnios, particularly beore midpregnancy, portends pulmonary hypoplasia and poor prognosis. Te kidneys may initially maniest severe hydroureteronephrosis but later develop cystic renal dysplasia or become small and echogenic.
Outcome may be poor even with normal amnionic uid volume. Associated anomalies occur in 40 percent o cases, and aneuploidy has been reported in 5 to 8 percent (Hayden, 1988; Hobbins, 1984; Mann, 2010). Evaluation and potential etal therapy or bladder outlet obstruction is discussed in Chapter 19 (p. 376).
SKELETON
Te standard examination includes demonstrating the presence o the arms, legs, hands, and eet (American Institute o Ultrasound in Medicine, 2018). Te detailed examination includes, in addition, the number and position o the digits—ngers and toes (Fig. 15-62). Te Nosology and Classication o Genetic Skeletal Disorders includes 461 skeletal anomalies in 42 groups, characterized according to their molecular phenotype, clinical eatures, or radiographic ndings (Mortier, 2019). More than 90 percent o skeletal disorders now have a known genetic basis, with pathogenic variants identied in more than 400 genes. Te two types o skeletal dysplasias are osteochondrodysplasias—the generalized abnormal development o bone and/or cartilage, and dysostoses—which are abnormalities o individual bones. In addition to these malormations, skeletal abnormalities include deormations, as with some cases o club- oot, and disruptions such as limb-reduction deects.
■ Skeletal Dysplasias
Te prevalence o skeletal dysplasias approximates 3 in 10,000 births. wo groups account or more than hal o all cases: the broblast growth actor 3 (FGFR3) chondrodysplasia group and the osteogenesis imperecta and decreased bone density group. Each occurs in 0.8 in 10,000 births (Stevenson, 2012). Evaluation o a pregnancy with suspected skeletal dysplasia includes a survey o every long bone, as well as the hands and eet, skull size and shape, clavicles, scapulae, thorax, and spine. Reerence tables are used to determine which long bones are aected and ascertain the degree o shortening (Appendix, p. 1239). Involvement o all long bones is termed micromelia, whereas predominant involvement o only the proximal, intermediate, or distal long bone segments is termed rhizomelia, mesomelia, and acromelia, respectively. Te degree o ossication should be noted, as should presence o bowing or ractures. Although precise characterization may elude prenatal diagnosis, it is requently possible to determine whether a skeletal dysplasia is lethal. Lethal dysplasias show proound long bone
A B
FIGURE 15-60 Autosomal recessive polycystic kidney disease (ARPKD). Transverse (A) and coronal (B) images of a 29-week fetus with marked renal enlargement. The kidneys measure 7 cm in length and have a ground-glass appearance. There is anhydramnios.
A
B
K
K
B C
FIGURE 15-61 Bladder outlet obstruction. A. The bladder (B) is so large that it fills the abdomen in this 13-week fetus. B. By 18 weeks, the kidneys (K) have become brightly echogenic. C. The bladder is markedly dilated and thick-walled, with dilation of the proximal urethra, termed the “keyhole” sign (arrow).
shortening, oten with measurements ar below the 5th percentile, and display emur length-to-abdominal circumerence ratios <16 percent (Nelson, 2014; Rahemtullah, 1997; Ramus, 1998). Other sonographic abnormalities are generally evident. Pulmonary hypoplasia is suggested by a thoracic circumerence <80 percent o the abdominal circumerence value, a thoracic circumerence <2.5th percentile, and a cardiac circumerence >50 percent o the thoracic circumerence value (Appendix, p. 1238). Aected pregnancies may also develop hydramnios and/or hydrops.
Te FGFR3 chondrodysplasias include achondroplasia, hypochondroplasia, and thanatophoric dysplasia. Achondroplasia is the most common nonlethal skeletal dysplasia. An impressive 98 percent o cases are due to a specic point mutation in the FGFR3 gene.
Inheritance is autosomal dominant, and 80 percent o cases result rom a new mutation. Achondroplasia is characterized by long bone shortening that is predominantly rhizomelic, an enlarged head with rontal bossing, depressed nasal bridge, exaggerated lumbar lordosis, and a trident conguration o the hands. Intelligence is usually normal. Te emur and humerus measurements may not all below the 5th percentile until the early third trimester. In homozygotes, who represent 25 percent o the ospring o heterozygous parents, the condition is characterized by greater long bone shortening and is lethal.
Te other major class o FGFR3 dysplasias, thanatophoric dysplasia, is the most common lethal skeletal disorder. It is characterized by severe micromelia, and aected etuses— particularly those with type II—may develop a characteristic cloverlea skull deormity (Kleeblattschädel) due to craniosynostosis. Genetic testing is con- rmatory.
Osteogenesis imperecta represents a group o skeletal dysplasias typied by hypomineralization. Tere are multiple types, and more than 90 percent o cases are characterized by a mutation in the COL1A1 or COL1A2 gene. ype II, also called the perinatal orm, is lethal. Te skull displays a proound lack o ossication, and gentle pressure on the maternal abdomen rom the ultrasound transducer results in visible skull deormation (Fig. 15-63). Other eatures include multiple in-utero ractures and ribs that appear “beaded.” Inheritance is autosomal dominant, such that all cases result rom either new mutations or gonadal mosaicism. Another skeletal dysplasia that creates severe hypomineralization is hypophosphatasia, which has an autosomal recessive inheritance.
■ Polydactyly
Te most common skeletal abnormality is polydactyly, which occurs in approximately 1 in 1000 births. It is post-axial i on the side o the ulna or bula and pre-axial i on the side o the
A B
C D
FIGURE 15-62 Normal extremities. A. Footprint with toes identified. B. Normal ankle position. C. Hand with fingers identified. D. Normal hand and forearm position.
A B
FIGURE 15-63 Post-axial polydactyly. A. In this image of the hand, the arrow points to a rudimentary digit adjacent to the little finger. B. Foot with 6 toes in a fetus with trisomy 13. The arrow points to the extra digit.
radius or tibia (Fig. 15-64). Te extra digit is oten rudimentary, and in the absence o a bony component, prenatal detection may be limited. Post-axial polydactyly is more common and is requently inherited in an autosomal dominant ashion.
Polydactyly is also a eature o syndromes such as Meckel-Gruber and trisomy 13.
■ Clubfoot—Talipes Equinovarus
Tis disorder is notable or a deormed talus and shortened Achilles tendon. Te aected oot is abnormally xed and positioned with equinus (downward pointing), varus (inward rotation), and oreoot adduction. Most cases are considered malormations, with a multiactorial genetic component. However, an association with environmental actors and with early amniocentesis suggests that deormation also plays a role (redwell, 2001). Sonographically, the ootprint is visible in the same plane as the tibia and bula (Fig. 15-65).
Te prevalence o cluboot approximates 1 in 1000 births, and the male:emale ratio is 2:1 (Carey, 2003; Pavone, 2012). Cluboot is bilateral in approximately 50 percent o cases. At least 50 percent o aected individuals have associated abnormalities, such as neural-tube deects, arthrogryposis, and myotonic dystrophy and other genetic syndromes (Mammen, 2004; Sharma, 2011). I there are other abnormalities, aneuploidy is present in 30 percent. In contrast, the aneuploidy rate is <4 percent when club- oot appears isolated (Lauson, 2010; Sharma, 2011). Tus, a careul search or associated abnormalities is warranted, and chromosomal microarray analysis may be considered.
■ LimbReduction Defects
Documentation o the arms and legs is a component o the standard examination. Te absence or hypoplasia o all or part o one or more extremities is a limb-reduction deect. Te birth prevalence is 4 to 8 in 10,000 (Kucik, 2012; Stoll, 2010; Vasluian, 2013).
Approximately hal o these are isolated deects, up to one third occur as part o a recognized syndrome, and individuals in the remaining cases have other coexisting anomalies (Stoll, 2010; Vasluian, 2013). Upper extremities are aected more requently than lower ones. A terminal transverse limb deect lacks part or all o a distal limb to create a stump (Fig. 15-66). Tis is more common than a longitudinal deect, which is complete or partial absence o the long bone(s) on only one side o a given extremity.
Absence o an entire extremity is termed amelia. Phocomelia, associated with thalidomide exposure, is an absence o one or more long bones with the hands or eet attached to the trunk (Chap. 8, p. 155). Limb-reduction deects are associated with numerous genetic syndromes, such
A B
C D
FIGURE 15-64 Osteogenesis imperfecta, Type IIa. A. Due to lack of skull ossification, gentle ultrasound transducer pressure on the maternal abdomen results in visible deformation (flattening) of the skull in this 24-week fetus. B. When the transducer pressure is removed, the skull shape returns to normal. C. In the four-chamber view of the heart, in-utero fractures lead to abrupt angulation of the ribs (arrows). D. The thorax is markedly smaller than the abdomen in this sagittal image.
FIGURE 15-65 Talipes equinovarus or clubfoot. This condition is diagnosed by visualizing the “footprint” in the same plane as the tibia and fibula.
as Roberts syndrome, an autosomal recessive condition characterized by tetraphocomelia. A clubhand deormity, usually rom an absent radius, is a component o the thrombocytopeniaabsent radius syndrome and is also associated with trisomy 18 (Fig. 16-5, p. 312). Limb-reduction deects may occur in the setting o a disruption such as amnionic-band sequence (Chap. 6, p. 113). Tey have also been associated with chorionic villus sampling when perormed beore 10 weeks’ gestation (Fig. 17-5, p. 346).
FIGURE 15-66 Transverse limb-reduction defect. A. At 18 weeks’ gestation, only a rudimentary hand was visible. B. By 24 weeks, the radius and ulna were normal in size and appearance, and small rudimentary digits were evident.
Nhận xét
Đăng nhận xét