Chapter 89. Achondroplasia
Key Points
■ Most common form of short-limbed dwarfism.
■ Incidence is 1 in 26,000 livebirths.
■ Most consistent sonographic finding is shortening of long bones between 21 and 27 weeks. Additional findings include macrocrania, frontal bossing, trident-shaped hand.
■ Differential diagnosis includes diastrophic dysplasia, achondrogenesis, Ellis–van Creveld syndrome, hypochondroplasia.
■ Condition is due to mutations in fibroblast growth factor receptor 3 (FGFR3) gene, which is a negative regulator of chondrocyte proliferation. Mutations activate the receptor and cause gain of function.
■ Pregnant women affected by achondroplasia need baseline pulmonary function tests and cesarean section delivery.
■ Prenatal diagnosis can be performed by sonography or by DNA analysis.
■ Major pediatric complications include short stature, foramen magnum compression, hydrocephalus, spinal stenosis, restrictive pulmonary disease, hypotonia, and recurrent ear infections. IQ is normal.
■ Inherited as an autosomal dominant condition, but 80% of cases are new mutations that always derive
from the father and are associated with advanced paternal age.
CONDITION
Achondroplasia is the most common form of short-limbed dwarfism. The condition has been recognized since ancient times. Dwarfs were accepted socially in ancient Egypt, and their daily activities, recorded through art, suggest not only assimilation into daily life, but also in some cases, a highranking position in society (Kozma, 2006). The term achondroplasia, meaning total absence of cartilage, was first used by Parrot in 1878 (Scott, 1976). Although this is not correct in a pathologic sense, the designation is commonly accepted.
INCIDENCE
The incidence of achondroplasia is 1 in 26,000 livebirths (Oberklaid et al., 1979). Earlierstudies that indicated an incidence of as high as 1 in 10,000 births probably included other causes of short-limbed dwarfism. More than 80% of cases are duetonewmutations (Shiang et al., 1994).Advanced paternal age was initially thought to be correlated with an increased incidence of new mutations resulting in achondroplasia (Murdoch et al., 1970). However, more recent observations have led to the alternative theory that sperms bearing the fibroblast growth factor receptor 3 (FGFR3) mutation that causes achondroplasia have aselective advantage over sperms without this mutation (Horton et al., 2007).
SONOGRAPHIC FINDINGS
Virtually all of the bones in the body are affected by
achondroplasia. Postnatal radiographic studies of the lumbar
spine, pelvis, and cranial regions permit definitive diagnosis

(Figure 89-1). Antenatally, diagnosis is complicated by a relatively normal appearance until the early second trimester.
The most consistent sonographic finding is shortening of the long bones, particularly the femur, occurring between 21 and 27 weeks of gestation (Figure 89-2) (Kurtz et al., 1986). The overall shape of the femurs is within normal limits. Initially, a Figure 89-1 Postnatal radiograph of a patientwith achondroplasia demonstrating flattened vertebral bodies with increased intervertebral space, cupped anterior ends to ribs, and hypoplastic midfacial bones. (Reprinted, with permission, from Cordone M, Lituania M, Bocchino G, Passamonti U, Toma P, Camera G. Ultrasonographic features in a case of heterozygous achondroplasia at 25 weeks’ gestation. Prenat Diagn. 1993;13:400. Copyright 1993 John Wiley & Sons, Ltd. Reprinted, with permission, of John Wiley & Sons, Ltd.)

Figure 89-2 Shortening (<5%) and bowing of the femur in a fetus at 29 weeks of gestation with heterozygous achondroplasia. normal relationship ofbiparietal diameterto femurispresent, but these measurements become progressively asynchronous over time (Filly et al., 1981). Additional findings that have been described during thesecondtrimesterinclude large head (macrocrania), abnormal facial profile due to frontal bossing (Figure 89-3), protuberant abdomen, and trident-shaped hand (Figure 89-4) (Cordone et al., 1993).

Figure 89-3 Antenatal facial profile of the fetus in Figure 89-4 and the infant in Figure 89-1, demonstrating frontal bossing, depressed nasal bridge, and an elongated philtrum. (Reprinted, with permission, from Cordone M, Lituania M, Bocchino G, Passamonti U, Toma P, Camera G. Ultrasonographic features in a case of heterozygous achondroplasia at 25 weeks’ gestation. Prenat Diagn. 1993;13:398. Copyright 1993 John Wiley & Sons, Ltd. Reprinted, with permission, of John Wiley & Sons, Ltd.)610 Part II Management of Fetal Conditions Diagnosed by Sonography

Figure 89-4 Prenatal sonogram of fetal hand at 25 weeks of gestation demonstrating relatively short phalanges and tridentlike appearance. (Reprinted, with permission, from Cordone M, Lituania M, Bocchino G, Passamonti U, Toma P, Camera G. Ultrasonographic features in a case of heterozygous achondroplasia at 25 weeks’ gestation. Prenat Diagn. 1993;13:397. Copyright 1993 John Wiley & Sons, Ltd. Reprinted, with permission, of John Wiley & Sons, Ltd.)
For prenatal diagnosis in the setting of one parent affected with achondroplasia, the fetus is considered to be affected if the length of the long bones is less than the third percentile or if polyhydramnios is present (Lattanzi and Harger, 1982; Elejalde et al., 1983). If both parents are affected by achondroplasia, the fetus is at 25% risk of inheriting both mutant alleles. In a retrospective review of 15 fetuses at 25% risk of homozygous achondroplasia, Patel and Filly (1995) demonstrated that fetal femoral length dropped below the third percentile at a mean of 15.6 weeks and 21.5 weeks in fetuses with homozygous and heterozygous achondroplasia, respectively.
If both parents are unaffected, prenatal diagnosis is more challenging. A fetus in which long bone growth is initially normal but then drops below the 10th percentile during the third trimester needs to be serially evaluated for the possibility of achondroplasia or hypochondroplasia. Hypochondroplasia is considered to be an allele of achondroplasia, with less severe clinical manifestations. Krakow et al. (2003) compared 2D and 3D imaging in the diagnosis of skeletal anomalies. In a case of achondroplasia, 3D imaging captured the trident hands and more clearly delineated the disproportionate aspects of the limbs.
For example, the left arm raised to the fetal forehead demonstrated that the level of the elbow was at the chin instead of the nose due to foreshortening of the proximal part of the arm (rhizomelia).
In one case report, an increased nuchal translucency measurement was noted in a fetus that was subsequently confirmed by molecular diagnosis to have achondroplasia. Rhizomelia, narrow thorax, and macrocrania were not observed until 18 weeks (Tonni et al., 2005).
DIFFERENTIAL DIAGNOSIS
The most likely diagnosis for a fetus with shortened long bones is that the fetus is normal or has intrauterine growth restriction that is not due to a skeletal dysplasia. However, the differential diagnosis includes chromosomal abnormalities as well as other types of dwarfism, some of which may be lethal at birth. An important prenatal finding that distinguishes heterozygous achondroplasia from some of the other skeletal dysplasias is the initially normal first and second trimester long bone measurements.
Other considerations in the differential diagnosis include diastrophic dysplasia, a recessively inherited form of short-limbed dwarfism, with the additional findings of thickening of the external ear and the characteristic “hitch-hiker” thumbs (see Chapter 93). In achondrogenesis, a lethal, recessively inherited condition, there is deficient ossification of the vertebral bodies. As compared with achondroplasia, a greater discrepancy between head size and trunk exists in achondrogenesis (see Chapter 97). In chondroectodermal dysplasia (Ellis–van Creveld syndrome), progressive distal shortening of the extremities and postaxial polydactyly are present (see Chapter 94). In addition, congenital heart malformations are present in 50% of cases. In hypochondroplasia, the major findings are short stature and an increased upper-to-lower body segment ratio. The facial features are within normal limits.
ANTENATAL NATURAL HISTORY
Achondroplasia is due to mutations in the fibroblast growth factor receptor 3 (FGFR3) gene. FGFR3 is a negative regulator of chondrocyte proliferation and differentiation in the growth plate. Mutations in the gene therefore activate the receptor and cause a gain of function (Horton, 2006). They predominantly affect bones that develop by endochondral ossification (Kurtz et al., 1986; Horton, 2006). Membranous bone formation occurs at a normal rate (Murdoch et al., 1970).
The abnormality in achondroplasia is confined to cartilage, and consistsof a failureofinterstitial cellstoproliferate.Bones that are initially formed from cartilage,such as the long bones of the extremities, bones at the base of theskull, and vertebral bodies are affected by this condition.
In histologic studies of bone and cartilage from patients with achondroplasia, morphology is normal (Rimoin et al., 1976). The arrangement of rows and columns of cells is regular and well organized. The rate of endochondral ossification is reduced and this contrasts with the normal periosteal ossification. This disparity results in periosteal bone extending beyond the growth plate and gives the appearance of short squat bones with cupped ends. Ultrastructural studies are also generally normal. The only abnormalities demonstrated have been a relative increase in the number of dead cells surrounded by microscars that contain focal aggregations of collagen fibrils (Rimoin et al., 1976).611 Chapter 89 Achondroplasia
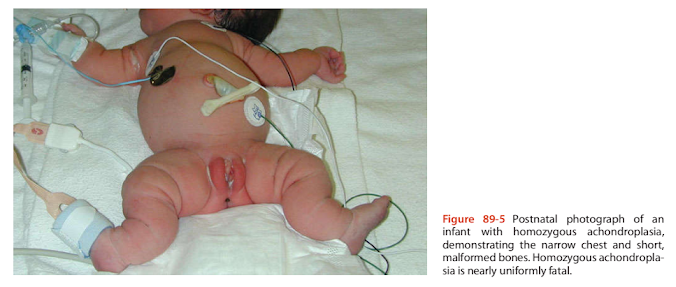
Figure 89-5 Postnatal photograph of an infant with homozygous achondroplasia, demonstrating the narrow chest and short, malformed bones. Homozygous achondroplasia is nearly uniformly fatal.
MANAGEMENT OF PREGNANCY
For an unaffected pregnant woman, the antenatal course for this condition is usually benign. When achondroplasia is suspected, serial sonography may be useful to determine if macrocrania is developing, which may necessitate cesarean delivery.
For the pregnant woman affected with achondroplasia, special problems include increased incidence of fetal loss,preeclampsia, and respiratory compromise in the third trimester (Trotter et al., 2005). Baseline pulmonary function studies should be performed. Cesarean delivery is mandatory for cephalopelvic disproportionsecondaryto marked pelvic contracture (Lattanzi andHarger, 1982;Allanson andHall, 1986).
The identification of the gene responsible for achondroplasia, FGFR3 (Shiang et al., 1994), allows DNA-based prenatal diagnosis. This is especially important for couples in which both partners are affected. In this situation, there is a 25% chance that the fetus will inherit the two mutant genes, resulting in homozygous achondroplasia that is associated with a very high incidence of fetal and neonatal death (Figure 89-5). In one report, first trimester DNA diagnosis has been described in a couple at risk for homozygous achondroplasia (Bellus et al., 1994).
In recent years, fetal chromosome analysis has been increasingly performed for shortfetal bones due tothe concerns of possible trisomy 21 (see Chapter 131). In thesetting of a fetus with short bones and a normal karyotype, amniotic fluid cells may be used as a source of fetal DNA to test for FGFR3 mutations. If an FGFR3 mutation is found, this is diagnostic.
FETAL INTERVENTION
There is no fetal intervention for achondroplasia.
TREATMENT OF THE NEWBORN
Most newborn infants with achondroplasia do not require special medical treatment and can be cared for in the regular nursery. In fact, many cases of achondroplasia are missed at birth. According to one study, even at 1 year of age, only 60% of cases are diagnosed (Saleh and Burton, 1991). Due to increased use of prenatalsonography in more recent studies it is estimated that approximately 20% of cases are missed at birth (Horton, 2006). For infants with achondroplasia, the main issues are definitive diagnosis and coordination of subspecialty care. All infants with a suspected diagnosis of achondroplasia should have radiographs taken of their long bones.
The salient clinical findings of achondroplasia in the newborn include short stature of the rhizomelic type. This means that the proximal arms and legs are relatively more shortened than the distal segments of the extremities. However, the short stature may be mild or even absent. Horton et al. (1978) showed that the range of birth length in newborns with achondroplasia overlaps the normal population.
Mean growth velocity is also normal during the first year of life. It is after the first year that growth velocity drops significantly. Other features of achondroplasia during the newborn period may include macrocrania, frontal bossing, and midface hypoplasia with coarsened facial features. The lumbar lordosis that is apparent later in life is rarely appreciated at birth. Cognitive development, socialization, speech, and language milestones are achieved normally, but initial gross motor milestones may be delayed (Scott, 1976). The muscular hypotonia responsible for delayed motor development disappears spontaneously at 4 to 6 years of age. The general pediatric recommendations for health supervision for children with achondroplasia have been summarized and updated (Trotter et al., 2005).
A serious potential problem in achondroplasia is cervicomedullary junction compression due to a small foramen612 Part II Management of Fetal Conditions Diagnosed by Sonography magnum. The foramen magnum is narrowed in a transverse direction (Hecht et al., 1989). This can result in occipitocervical pain, ataxia, incontinence, or the more serious complication of apnea and sudden infant death (Pauli et al., 1984). Magnetic resonance imaging (MRI) studies in five affected patients have demonstrated the discrepancy between the size of the brainstem and the foramen magnum (Thomas et al., 1988). In addition, hydrocephalus is frequently found due to anatomic abnormalities in the occipital bone that produce intracranial venous hypertension. It has been hypothesized that obstruction at the jugular foramen elevates intracranial venous pressure, resulting in decreased absorption of cerebrospinal fluid into the sagittal sinus, producing a communicating hydrocephalus (Thomas et al., 1988). Approximately 5% of affected children require placement of a shunt (Haga, 2004).
Approximately 10% of patients with achondroplasia have respiratory complications due to foramen magnum compression(Stokes et al., 1983;Thomas etal.,1988).Itis recommended that computed tomographic (CT) or MRI be performed in conjunction with somatosensory evoked potentials to screen for this problem (Reid et al., 1987). Although posterior fossa decompression and atlantal laminectomy represent major surgical intervention,symptoms have been reported to improve postoperatively (Ryken and Menezes, 1994). The increased mortality seen in achondroplasia (Hecht et al., 1987) is due to this brainstem compression and it primarily affects children younger than 4 years old.
SURGICAL TREATMENT
Cervicomedullary compressive surgery is indicated for individuals with symptoms of foramen magnum compression (Ho et al., 2004). This surgery can treat central apnea that may result in death, and/or relieve neurologic complications secondary to spinal cord damage. In one study, the quality of life and morbidity of individuals with achondroplasia who underwent cervicomedullary decompression surgery was indistinguishable from age and gender matched controls (Ho et al., 2004).
The orthopedic deformities in achondroplasia consist of bowing of the legs, hyperlordosis, and spinal stenosis. In approximately 25% of cases, the bowing of the legs is treated with corrective osteotomies of the tibia. This surgery is usually performed between 3 and 10 years of age.Althoughspinal stenosis is universal, only in relatively few cases does it cause cauda equina syndrome, which manifests as a lower motor neuron flaccid paralysis. This becomes evident during very late adolescence or adulthood and requires a decompressive laminectomy. The most severe but least common orthopedic problem is thoracolumbar kyphosis, which can cause paraplegia earlier in childhood. This requires aggressive spinal surgery.
Surgical therapies currently being optimized may also significantly improve the cosmetic aspects of achondroplasia.
Craniofacial surgery is possible to advance midfacial bones and correct severe dental malocclusion (Denny et al., 1992).
Leg lengtheninghas received much attention as a means of improving height and appearance for patients with achondroplasia (Figure 89-6). This has been utilized more commonly outside of North America. The surgery is painful; it should be performed on only adolescents; and it may take up to 2 years to complete (Saleh and Burton, 1991). Aldegheri and Dall’Oca (2001) reviewed their experience in performing limb lengthening on 80 achondroplastic individuals. Ten of these individuals also underwent humeral lengthening to improve ability to independently attend to matters of personal

Figure 89-6 Patient with achondroplasia demonstrating cosmetic effects of multiple leg-lengthening operations over an almost 4-year period. (Courtesy of Professor Michael Saleh, Sheffield Children’s Hospital, Sheffield, United Kingdom.)
hygiene. Patients gained an average of 20.5 cm in height but 39% of them had complications. The treatmenttime required was approximately 33 months. Limb lengthening surgery is invasive, complex, and potentially dangerous.
LONG-TERM OUTCOME
The long-term consequences of achondroplasia are cervicomedullary compression, hydrocephalus,spinal stenosis, restrictive and obstructive lung disease, otitis media, and thoracolumbar kyphosis (Hunter et al., 1998; Trotter et al., 2005).
Mental development is generally normal.
Standard growth curves exist to assess normal growth for children with achondroplasia (Horton et al., 1978). The average adult height for an affected male is 52 inches (129 cm) and an affected female 48.6 inches (122 cm). The average adult weight for a male is 120 lb (55kg) and a female 100 lb (45kg) (Murdoch et al., 1970; Scott, 1976). Obesity is common (Hecht et al., 1988). In sporadic cases, parental height does not significantly influence the adult height of affected offspring (Murdoch et al., 1970; Scott, 1976).
In an initial clinical trial, recombinant human growth hormone given to patients with achondroplasia at doses of 0.3 mg per kilogram of body weight per week moderately increased overall height velocity, particularly in the short term. There were no untoward effects, particularly with regard to worsening of the degree of spinal stenosis (Horton et al., 1992). Further clinical trials have not shown a clear long-term benefit. Currently, most experts do not recommend growth hormone for achondroplasia (Horton et al., 2007). Further medical problems for children with achondroplasiaincluderecurrent ear infections (in90%of children) and conductive and sensorineural hearing loss. Patients with achondroplasia have specific structural changes of the temporal bone, although it is unclear how these changes relate to hearing loss (Shohat et al., 1993). Approximately half of the affected children undergo placement of ventilation tubes in the tympanic membrane (Haga, 2004). For many patients, dental crowding necessitates orthodontic treatment.
Intelligence quotient (IQ) is normal for individuals with achondroplasia. One study compared 20 adults affected with achondroplasia with siblings of the same sex and found that the mean number of years of formal education was comparable for each group. Females, more than males, have a significantly lower occupational level as compared with their same-sex siblings (Roizen et al., 1990). It was questioned whether this was due to a self-esteem problem that was more profound in females. The psychiatric aspects of achondroplasia have also been studied. In general, patients with achondroplasia had achieved satisfactory life adjustment and had secure identities as “little people” (Brust et al., 1976). The functional health status of adults with achondroplasia is not drastically reduced in comparison with the general United
States population (Mahomed et al., 1998).
Looking toward the future, novel therapies will be directed toward countering the effects of the overactive FGFR3 (Horton, 2006, Horton et al., 2007). Chemical inhibitors that downregulate the tyrosine kinase activity of FGFR3 might be used in a manner analogous to the treatment of chronic lymphocytic leukemia with inhibitors to Bcr-abl tyrosine kinase. Alternatively, antibodies could be used to interfere with the binding of FGF ligands to FGFR3. The molecular mechanism responsible for achondroplasia is the same as in many cancers. Also, C-type natriuretic peptide (CNP) downregulates FGF-induced activation of MAP kinase signaling pathways in growth plate chondrocytes. It has been suggested as a treatment for achondroplasia because of therapeutic effects observed in mouse models (Horton, 2006).
GENETICS AND RECURRENCE RISK
Achondroplasia is inherited as an autosomal dominant condition. Fifty percent of an affected parent’s offspring will also have achondroplasia. The phenotype is 100% penetrant, which means that all offspring who carry the gene will express the full clinical appearance of achondroplasia. If both parents are affected, there is a 50% chance of having a child with heterozygous achondroplasia, a 25% chance of having a child with homozygous achondroplasia, and a 25% chance of having a child with normal stature.
As stated earlier, the majority of cases (80%) are due to new mutations. Murdoch et al. (1970) studied 148 patients with achondroplasia. Thirty-one (21%) had one or both parents affected, whereas 117 cases (79%) had no family history of the condition. Although a case has been described of one family in which normal parents gave birth to multiple affected children (Fryns et al., 1983), a larger Canadian study that examined the risk of recurrence due to parental gonadal mosaicism found only 1 case in 443 full siblings of children with achondroplasia (Mettler and Fraser, 2000). Thus the recurrence risk if the parents are normal is only 0.02%.
The gene involved in achondroplasia was localized to chromosome 4 band p16.3 in 1994 (LeMerrer et al., 1994). The disease is caused by mutations in the coding sequences for FGFR3. At least nine different heparin-binding fibroblast growth factors are known.All of them have pleiotropic effects on different cell types and have quite different patterns of expression during development. Evidence that the FGFR3 gene was a good “candidate” gene for achondroplasia included the fact that FGFR3 mRNA is found in the central nervous system and all of the prebone cartilage structures of the mouse (Shiang et al., 1994).
In a landmark study, Shiang et al. (1994) demonstrated point mutations in the DNA of 15 of 16 individuals studied with achondroplasia. The mutations all occurred at nucleotide 1138, and resulted in the substitution of an arginine residue for a glycine at position 380 of the mature protein, which is in the transmembrane domain of FGFR3. This work has been validated and extended by other investigators, who have also shown that all achondroplasia mutations studied result in the same substitutions at the same amino acid of the transmembrane domain of the FGFR3 protein614
Part II Management of Fetal Conditions Diagnosed by Sonography
(Bellus et al., 1994; Rousseau et al., 1994). The homogeneousnatureofthesemutationsin achondroplasiaisunprecedented for an autosomal dominantdisorder. This may explain the phenotypic similarity between all patients with achondroplasia. FGFR3 has subsequently been shown to be one of the most mutable genes in the human genome (Vajo et al., 2000).
Based on these results, rapid polymerase chain reaction-based prenatal diagnosis is available for the condition (Bellus et al., 1994). DNA testing is relatively straightforward because the mutations involved are minimal in number, and they create new recognition sites for restriction endonucleases (Francomano, 1995). In sporadic cases of achondroplasia, the FGFR3 mutation arisesduringspermatogenesis and is associated with advanced paternal age (Wilkin et al., 1998; Vajo et al., 2000).
It is now known that achondroplasia is one of six diseases caused by mutations in the FGFR3 gene. Four are associated with skeletal dysplasias and two are associated with craniosynostosis. The other three skeletal dysplasias are hypochondroplasia, thanatophoric dysplasia (see Chapter 90), and severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) (Bonaventure et al., 1996;Bellus et al., 1999). FGFR3 mutations also causeMuenke coronal craniosynostosis and Crouzon syndrome with acanthosis nigricans (Vajo et al., 2000) (see Chapter 10)
Nhận xét
Đăng nhận xét